金及二元合金团簇吸附小分子的第一性原理研究
2014-07-06王雪方王新强
王雪方,王新强
(重庆大学物理学院,重庆 401331)
自Haruta[1]等发现纳米尺寸的金团簇对CO具有较高的低温氧化活性以来,金纳米团簇的催化氧化机理受到了众多科研工作者的关注。目前,大多数理论研究都集中在小尺寸金团簇上[2],而对大尺寸的金团簇上小分子的吸附研究较少,事实上金团簇的尺寸越大越接近实际应用。另外,由2种元素组成的二元金属纳米团簇比单一的金属纳米团簇受到了更为广泛的关注[3-4],这主要因为某些特定的金属原子掺杂在金团簇中极大地提高了金团簇的催化效果。一般情况下,关于金团簇催化氧化性质的理论和实验研究都是在干燥的条件下进行的,然而某些在湿润条件下的研究结果[5-6]表明,水分子的加入会提高氧气的活性,促进 CO氧化。Okumura等[5]将 O2和 H2O分子共同吸附在Au10团簇上,发现H2O分子的加入提高了吸附在金团簇表面上的O2分子的活性。
有关小尺寸金团簇吸附小分子的研究较多,而关于大尺寸金团簇在掺杂和加水条件下吸附小分子的研究尚不全面。本文采用第一性原理方法研究了CO和O2在Au32及其掺杂团簇上的吸附,为下一步研究CO和O2在这些团簇上的氧化反应机理提供参考。在此基础上,深入研究O2和H2O在Au32和Au20Pd12团簇上的共吸附行为是理解Au团簇在潮气条件下催化活性的第一步。
1 计算方法
计算工作采用基于密度泛函理论的Dmol3软件包[7]完成。电子交换关联势采用基于广义梯度近似(GGA)的PW91泛函[8]。计算时选取相对论半芯贋势(A relativistic semi-core pseudopotential)[9]和双数值极化基组(DNP),未限制电子自旋。计算时优化构型的频率以确认没有虚频。计算精度设置为Medium,优化收敛精度取程序预设值。为了验证所选方法的合理性,在同等条件下计算了 Au2,CO和 O2的键长,分别为 0.253,0.114 和 0.122 nm,与实验值 0.247,0.113 和0.121 nm 一致[10-11]。
2 结果与讨论
Au32团簇为正二十面体结构,如图1(a)所示,含12个五配位和20个六配位的Au原子。将其五配位Au原子用Pd原子替代,优化后所得的Au20Pd12团簇仍然是正二十面体结构,如图1(b)所示。Au20Pd12团簇的平均结合能为1.14 eV,比Au32大0.41 eV,所以正二十面体结构的Au20Pd12团簇是稳定的。当小分子在Au32和Au20Pd12团簇上吸附时,其吸附点共有5个,分别是顶位T1,T2,桥位B1,B2和孔位H1,如图1(a)和(b)所示。计算过程中考虑了所有可能的吸附形式。

图1 Au32和Au20Pd12团簇优化后的构型及可能吸附点
2.1 CO在Au32和Au20Pd12团簇上的吸附
Au32团簇吸附CO的5种构型中,孔位H1的吸附结构不稳定,桥位B1的吸附结构优化后变为顶位T1,桥位B2的吸附结构不稳定,只有顶位T1,T2的吸附结构稳定,如图2(a)和(b)所示,相关参数见表1。Au20Pd12团簇吸附CO的5种构型中,孔位H1的吸附结构优化后变为桥位B1,桥位B2的吸附结构优化变为顶位T1,其他结构均经优化得到,如图2(c),(d)和(e)所示,相关参数见表1。

图2 Au32和Au20Pd12团簇吸附CO优化后的构型
定义CO的吸附能公式为:Eab=E[CO]+E[Au32or Au20Pd12]-E[Au32-CO or Au20Pd12-CO]。CO在Au32团簇的T1和T2位置的吸附能分别为1.32 和0.88 eV,Au-C 键长为 0.199 和0.208 nm,在T1位置吸附较稳定。CO在Au20Pd12团簇的T1,T2和B1位置的吸附能分别为2.30,1.47和0.57 eV,同样在T1位置吸附较稳定。对比CO在Au32和Au20Pd12团簇上的吸附发现:CO在Au32和Au20Pd12团簇的T1位置的吸附能分别为1.32 和2.30 eV,在 T2 位置的吸附能分别为0.88和1.47,表明在相同构型同种吸附位上,CO在掺杂团簇Au20Pd12上的吸附能比纯金团簇Au32大,吸附更稳定。CO在Au32和Au20Pd12团簇的T1位置吸附后,其带电量为 0.009和 0.272 e,键长为0.115 和0.116 nm,表明在 Au20Pd12团簇的 T1 吸附模式中,C-O键活化和电荷转移稍大。

表1 Au32和Au20Pd12团簇吸附CO的吸附能(Eab/eV)、CO上的电荷(QCO/e)、Au/Pd-C键长(LAu/Pd-C/nm)和C-O键长(LC-O/nm)
2.2 H2O分子的加入对 O 2在 A u32和 A u20Pd12团簇吸附的影响
2.2.1 O2在 Au32和 Au20Pd12团簇上的吸附
O2在Au32团簇上的吸附中,只有顶位T1吸附结构(Au32-O)稳定存在,如图3(a)所示。O2在Au20Pd12团簇上的吸附中,只有顶位T1和桥位B1的吸附结构(Au20Pd12-O和Au20Pd12-O)稳定存在,如图3(b)和(c)所示,相关参数见表2。吸附能的计算公式:Eab=E[O2]+E[Au32or Au20Pd12]-E[Au32-O2or Au20Pd12-O2]。O2在 Au32团簇的T1位置的吸附能为0.319 eV,O2在Au20Pd12团簇T1和B1位置的吸附能分别为0.533和0.725 eV,说明 O2在 Au20Pd12团簇上的吸附更稳定。
2.2.2 H2O和O2在Au32团簇表面的共吸附
考虑H2O分子的加入对O2在Au32团簇吸附的影响,对于Au32-OT12构型,让H2O分别吸附在近邻O2吸附位的五配位和六配位处,优化后得到团簇构型Au32-OT12-H2OT1和Au32-OT12-H2OT2,如图4(a)和(b)所示,相关参数见表3。吸附能的计算公式如下:Eab=E[O2]+E[H2O]+E[Au32or Au20Pd12]-E[Au32-O2-H2O or Au20Pd12-O2-H2O]。
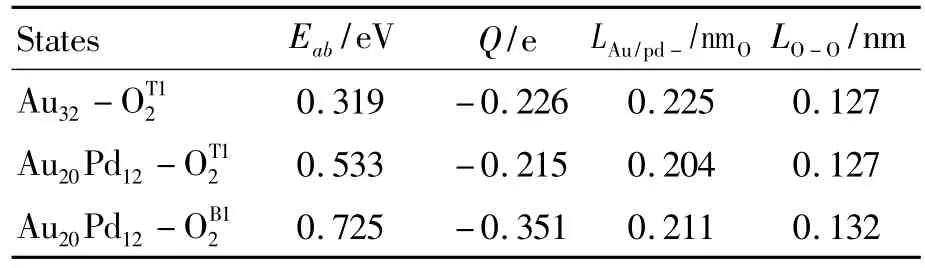
表2 Au32和Au20Pd12团簇吸附O2的吸附能(Eab/eV)、O2上的电荷(Q/e),Au/Pd-O键长(LAu/pd-O/nm)和O-O键长(LO-O/nm)

图3 Au32和Au20Pd12团簇吸附O2优化后的构型
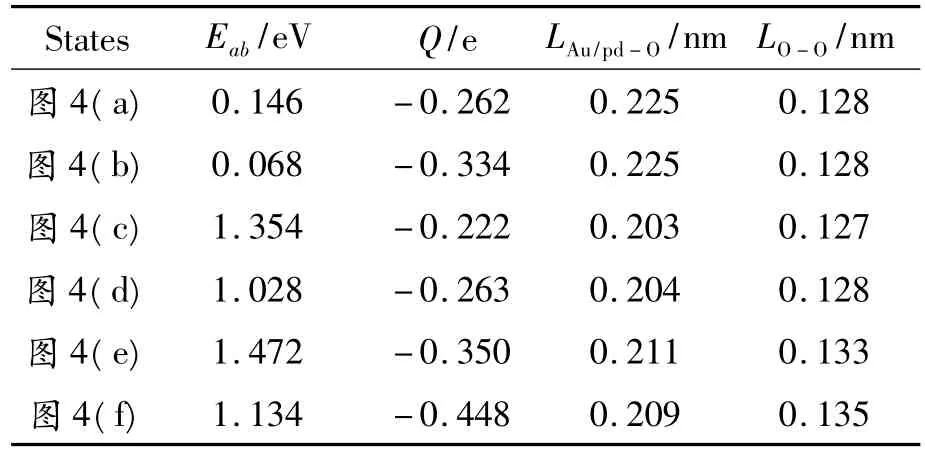
表3 O2和H2O在Au32和Au20Pd12团簇上共吸附的吸附能(Eab/eV)、O2上的电荷(Q/e)、Au/Pd-O键长(LAu/pd-O/nm)和O-O键长(LO-O/nm)

图4 O2和H2O在Au32和Au20Pd12团簇上共吸附优化后的构型
比较构型Au32--H2OT1和 Au32--H2OT2的吸附能,发现H2O和O2更容易共同吸附到Au32团簇的 T1位置。对比构型 Au32-、Au32--H2OT1和Au32--H2OT2中O-O键长和O2带电量发现,共吸附模型中的O-O键长比单独O2吸附模型中的O-O键长要长,且共吸附模型中O2的带电量稍大。这说明H2O能够促使O-O键活性增强,共吸附模式对O2分子活化程度较大。
考虑H2O分子的加入对O2在Au20Pd12团簇吸附的影响。对于Au20Pd12-和Au20Pd12-构型,令分别吸附在近邻吸附位的五配位和六配位处,优化后得到构型Au20Pd12--H2OT1,Au20Pd12--H2OT2,Au20Pd12--H2OT1和 A u20Pd12--H2OT2,如图 4 (c),(d),(e)和(f)所示,相关参数见表4。比较表3中相关数据发现:H2O同样能够使吸附在Au20Pd12团簇上的O2分子活性增强。
3 结论
通过以上研究可知:① CO和 O2在 Au32及Au20Pd12团簇上五配位处吸附最稳定,配位数越低,越有利于吸附;② 与Au32团簇相比,CO和O2更倾向于吸附在Au20Pd12团簇表面;③ H2O分子的加入能够提高吸附在Au32及Au20Pd12团簇表面的O2分子的活性,更有利于接下来催化反应的进行。
[1]Haruta M,Kobayashi T,Sano H,et al.NOVEL GOLD CATALYSTS FOR THE OXIDATION OF CARBONMONOXIDE AT A TEMPERATURE FAR BELOW 0-DEGREES-C[J].Chemistry Letters,1987(2):405-408.
[2]Wu X,Senapati L,Nayak S K,et al.A density functional study of carbon monoxide adsorption on small cationic,neutral,and anionic gold clusters[J].Journal of Chemical Physics,2002,117(8):4010-4015.
[3]Zhang F,Fa W.Doping golden clusters:MAu19-and M2Au18-(M=Cu and Na)[J].Physics Letters A,2012,376(19):1612-1616.
[4]Chang C M,Cheng C,Wei C M.CO oxidation on unsupported Au55,Ag55,and Au25Ag30 nanoclusters[J].The Journal of chemical,134l physics,2008,128(12):124710-124710.
[5]Okumura M,Haruta M,Kitagawa Y,et al.Theoretical study of H2O and O-2 adsorption on Au small clusters[J].Gold Bulletin,2007,40(1):40-44.
[6]Bongiorno A,Landman U.Water-enhanced catalysis of CO oxidation on free and supported gold nanoclusters[J].Physical Review Letters,2005,95(10).
[7]Delley B.AN ALL-ELECTRON NUMERICAL-METHOD FOR SOLVING THE LOCAL DENSITY FUNCTIONAL FOR POLYATOMIC-MOLECULES[J].Journal of Chemical Physics,1990,92(1):508-517.
[8]Perdew J P,Wang Y.ACCURATE AND SIMPLE ANALYTIC REPRESENTATION OF THE ELECTRON-GAS CORRELATION-ENERGY[J].PhysicalReviewB,1992,45(23):13244-13249.
[9]Delley B.Hardness conserving semilocal pseudopotentials[J].Physical Review B,2002,66(15).
[10]Weast R.Handbook of Chemistry and Physics 55th[M].Cleveland:CRC Press,1974.
[11]Wang Y,Gong X G.First-principles study of interaction of cluster Au-32 with CO,H-2,and O-2[J].Journal of Chemical Physics,2006,125(12).
