纤锌矿AlN中反位和间隙点缺陷电子结构的最大局域化Wannier函数方法研究
2014-06-27牛海波李冠强
牛海波, 李冠强
(1.西安交通大学城市学院 物理教学中心, 陕西 西安 710018; 2.陕西科技大学 理学院, 陕西 西安 710021)
0 引言
纤锌矿结构AlN是一种直接带隙宽禁带半导体材料,其带隙宽度达到6.2 eV,在Ⅲ-Ⅴ族半导体中禁带宽度最大.这一特点使其在深紫外发光器件领域有着重要的应用价值.
如Yoshitaka在2006年成功制作出了基于AlN的波长210 nm的LED[1];还通过组成AlGaN三元合金,可制成带宽可调的发光器件[2,3],这在环境保护、纳米制造、大容量存储等方面具有广阔的应用前景;此外,AlN晶体中由于[0001]晶向上结构存在中心不对称,产生较强的自发极化及压电极化,因此,在表面声波器件、压力传感器等方面也具有重要的应用价值.
点缺陷是晶体生长过程中最为常见的缺陷,它的存在会改变晶体的电子结构,进而对晶体光学性质产生重要影响.因此,有必要对点缺陷引起的电子结构变化进行研究.目前,对于AlN晶体中空位缺陷电子结构的研究报道较多,而对于间隙及反位缺陷的报道较少.
本文结合第一性原理计算,利用最大局域化Wannier函数对纤锌矿AlN中反位和间隙点缺陷的电子结构及化学成键进行了分析.研究发现相比于传统方法,最大局域化Wannier函数方法在分析缺陷电子结构上具有更加直观的特点,能给出清晰的物理图景.
1 计算理论及方法
1.1 最大局域化Wannier函数
晶体电子结构的计算一般采用的是倒空间中的周期性布洛赫函数|ψnk>.此外,也可以采用实空间的Wannier函数|Rn>,它可由布洛赫函数经过傅里叶变化得到,二者之间关系为:
(1)
其中,R为晶格参数,n为能带指数,V为晶胞体积,积分区间为整个布里渊区.由于布洛赫函数中的相位因子具有周期不确定性,导致式(1)得到的Wannier函数并不唯一,长久以来限制了它的应用.
为了解决这个问题,1997年,Marzari 和Vanderbilt提出了所谓最大局域化Wannier函数[4].即对Wannier函数在空间中的分布进行最大程度的局域化.其中局域化计算所依据的函数公式为:
(2)

为了加快局域化的过程,减少计算量,通常根据晶体的能带结构及原子轨道形状,首先对Wannier函数的形状及中心位置给出合理的初始值,再开始迭代计算.
与布洛赫函数呈现非局域性质不同,Wannier 函数在实空间中正交且局域分布.根据Wannier函数的形状及其中心位置分布,不仅可以便捷地分析晶体中的化学成键及键的离子性大小等性质[5,6],还适合用来分析晶体中缺陷的电子结构[7],计算晶体的介电性质、声光作用等性质;还通过Wannier函数插值可得到布里渊区中所要的对称线上的能带结构,从而利用Wannier 函数的局域性可构建基函数进行大尺度结构的电子结构计算,更多的应用可见文献[8].
1.2 计算方法
本文研究所用晶胞是基于AlN原胞建立的3×3×2的超晶胞,共含有72个原子,通过交换N、Al原子位置及在间隙位置放入相应原子来模拟反位和间隙点缺陷的情形.由于第一性原理计算中采用赝势来描述电子与离子实之间的相互作用,直接使用晶格参数的实验值一般会引入额外的力,造成体系能量上升,使得体系不稳定.因此,超晶胞的晶格参数由优化后的纤锌矿AlN 单胞晶格参数(a=3.116 Å,c=4.995 Å,u=0.381)来描述,与实验值(a=3.112 Å,c=4.982 Å,u=0.382)符合很好.
计算中采用基于密度泛函理论的平面波赝势方法,应用广义梯度近似(GGA)下的PBE来处理电子之间相互作用产生的交换关联能,电子-离子实之间的相互作用选择Vanderbilt超软赝势来描述,平面波截断能设为40 Ry,选取3×3×3 的Monkorst-Park K点对全布里渊区求和,总能变化收敛的标准为1.0×10-6eV,原子上的应力收敛标准为0.005 eV/Å.
以上计算利用第一性原理软件Quantum Espresso[9]进行.它是以密度泛函理论和分子动力学理论为基础的应用广泛的开源软件.最大局域化Wannier函数利用Wannier90程序包[10]进行计算,式(2)中Ω达到最大局域化的标准设为相邻两步迭代计算之间相差为1.0×10-8Å2.Wannier90作为一种后处理程序,和Quantum Espresso软件实现了很好的衔接.计算时,利用Quantum Espresso软件进行自洽及非自洽计算,可得到基态电子波函数,再由Wannier90程序进行后处理得到最大局域化Wannier函数.
2 计算结果及讨论
2.1 理想结构的电子态分析
为了后续更好地进行对比分析,我们首先对理想纤锌矿AlN结构中的最大局域化Wannier函数进行了计算,进而利用Wannier函数的形状及其中心分布分析了电子结构.
在计算过程中,由于N、Al原子轨道sp3杂化,因此,为了得到更合理的最大局域化Wannier函数,我们以sp3杂化轨道作为初始Wannier函数形状,以N原子位置为初始Wannier函数中心位置,以价带为一个整体计算了其对应的最大局域化Wannier函数;为了判断计算得到的最大局域化Wannier函数的合理性,我们将Wannier函数插值得到的价带与第一性原理计算得到的价带进行了比较,如图1(a)所示.可以看到二者符合非常好.此外,每个Wannier函数对应的Ω处在0.94~0.96 Å2之间,远小于Wannier90程序一般所要求的5 Å2的数值,这说明得到的最大局域化Wannier函数正确合理.
图1(b)给出了Wannier函数的形状,清晰地反映了AlN晶体中N-Al键sp3-sp3σ成键的特性.Wannier函数分布靠近N原子,说明N-Al键具有离子性.通过图1 (c)Wannier函数中心分布更能体现N-Al键这一特点.Wannier函数中心非常靠近N原子,说明N-Al键具有较强的离子性.根据Hazem[11]利用Wannier 函数中心位置来判断键的离子性的方法,我们计算得到的N-Al键的离子性为0.621,与文献[11]中给出的0.612的结果非常接近,在Ⅲ-Ⅴ族化合物中最高.

(a)第一性原理计算得到的理想AlN能带结构(黑线)与Wannier函数插值得到的价带(红色点线)

(b)Wannier函数形状 (c)Wannier函数中心分布图1 理想AlN能带结构、Wannier函数形状及中心分布
2.2 反位缺陷的电子结构分析
AlN中两种反位缺陷分别是N占据Al原子格点位置(NAl)及Al占据N原子格点位置(AlN).在两种缺陷结构优化基础上,计算得到的能带结构如图2所示.
由于悬挂键的相互作用,带隙中引入了缺陷态能带,在NAl能带结构中,两条缺陷态能带处在费米能级之下,被电子完全填充,另外两条缺陷态能带处在费米能级之上,形成了空带.因此,以价带及被电子完全填充的缺陷态能带为一个整体计算了最大局域化Wannier函数,共得到145个完全填充的Wannier函数.
在AlN能带结构中,一条缺陷态能带降入价带中,被电子完全填充,另外三条缺陷态能带发生简并且被费米能级穿越,产生部分填充.对于这种情况,我们以价带及全部缺陷态能带为一个整体计算了最大局域化Wannier函数,共得到144个部分占据的Wannier函数.
在以上两种情况下,通过将Wannier函数插值得到的能带与第一性原理计算得到的能带进行比较,发现两者符合非常好,这说明最大局域化Wannier函数计算合理.

(a)NAl缺陷结构能带图 (b)AlN缺陷结构能带图(黑线)图2 两种反位缺陷的能带结构,Wannier函数插值得到的能带图用红线表示
2.2.1 NAl电子结构分析
通过观察NAl结构中Wannier函数中心的分布,我们发现在反位缺陷次近邻以外的结构中,Wannier函数中心的分布与理想结构中一致,说明NAl对晶体电子结构的影响主要施加在次近邻以内的结构中.
图3(a)给出了NAl中次近邻以内结构中的Wannier中心分布,清晰地反映了NAl中电子结构的变化.可以看到,缺陷周围的Wannier函数中心向替代N原子产生了移动,这是由于N原子核对电子的吸引作用要强于Al原子核,当N替代Al原子后,将吸引电子云产生移动.[0001]方向上的N原子周围的Wannier函数中心的移动最大,这是因为[0001]方向上的N原子周围的Wannier函数中心距替代N原子最近,导致吸引作用更强,使得这些Wannier函数中心位置变化最剧烈.此外,还可以看到有一个Wannier函数中心非常靠近替代N原子,这是由于N替代Al原子后会多出2个电子,从而形成了孤对电子,且替代N原子核对其吸引力最强.
分析Wannier函数的形状可以得到原子间成键信息.图3(b)所示为沿[0001]方向上两相邻N原子间Wannier函数形状.这是典型的p-pσ键形状,反映了替代N原子与[0001]方向上的最近邻N原子成键,这与Stampfl[12]利用传统的第一性原理方法计算得到的结论相同,但是Wannier方法更直接.
在图3(c)中可以发现,替代N原子与垂直于[0001]方向底面上N原子间Wannier函数大部分局域在底面N原子附近,而不是居于两者之间,说明悬挂键间尽管有相互作用,但并没有成键,其结果只是形成孤对电子.文献[6]对同是Ⅲ-Ⅴ族氮化物的纤锌矿GaN中的反位N原子进行了研究,结果与本文一致.这说明这两种氮化物中的反位N原子具有相似的性质.以上分析体现了Wannier函数方法的直观性.

(a) NAl次近邻结构中Wannier函数中心分布,缺陷最近邻Wannier函数用红色小球表示 (b)[0001]方向上Wannier函数形状 (c)垂直于[0001]方向上底面上Wannier函数形状图3 NAl结构中Wannier函数形状及中心分布
2.2.2 AlN电子结构分析
与上述NAl结构情况相似,我们也发现在AlN结构中,次近邻以外的结构中Wannier函数中心分布与理想结构中完全一致,显示AlN缺陷对晶体电子结构的影响也局限在次近邻以内的结构中.
图4 (a) 给出了AlN中次近邻以内结构中的Wannier函数中心分布,与NAl结构中Wannier函数分布相比,可以看到次近邻N原子周围的Wannier函数位置变化更大,表明AlN对次近邻以内电子结构的影响要大于NAl.观察缺陷最近邻的四个Wannier函数中心的分布,发现在与[0001]方向垂直的底面上,三个Wannier函数中心分布在替代Al原子及近邻Al原子中间偏上的位置,这是由于Al替代N原子后,Al原子核对电子云的吸引较N原子核减弱,使得原来靠近N原子的电子云发生了移动.
Wannier函数中心并没有在替代Al原子及近邻Al原子几何连线上,且从图4(c)给出的对应的Wannier函数形状可以看到相邻Wannier函数有较大的重叠,其形状也为σ成键,说明相邻3个Al原子间悬挂键发生了较强作用并成键.[0001]方向上Wannier函数中心更靠近替代Al原子,表明孤对电子局域在替代Al原子附近,其对应Wannier函数形状由图4(b)给出.
2.3 间隙缺陷的电子结构分析
纤锌矿结构AlN中间隙位置有两种,分别为四面体间隙位置及八面体间隙位置.由于八面体间隙位置更稳定,本文以八面体间隙位置为对象,研究了N间隙(Ni)和Al间隙(Ali)两种缺陷的电子结构.在两种缺陷结构优化的基础上,利用第一性原理计算得到的能带结构如图5所示.
在Ni中,由于费米能级穿越缺陷态能带引起部分占据,而在Ali中,费米能级进入导带,三条缺陷态能带也进入到导带中发生耦合,在这两种缺陷结构中,我们对价带及全部缺陷态能带进行了最大局域化Wannier函数计算.为了评判计算得到的最大局域化Wannier函数的质量,通过Wannier函数插值得到的能带也在图5中给出.可以发现,在Ni中两种结构符合非常好,而在Ali中,大部分符合很好,但在能量最高的缺陷态能带处有部分的偏离.这是因为电子进入导带后,局域性减弱导致,这种小部分的偏移对最后结果的影响较小.
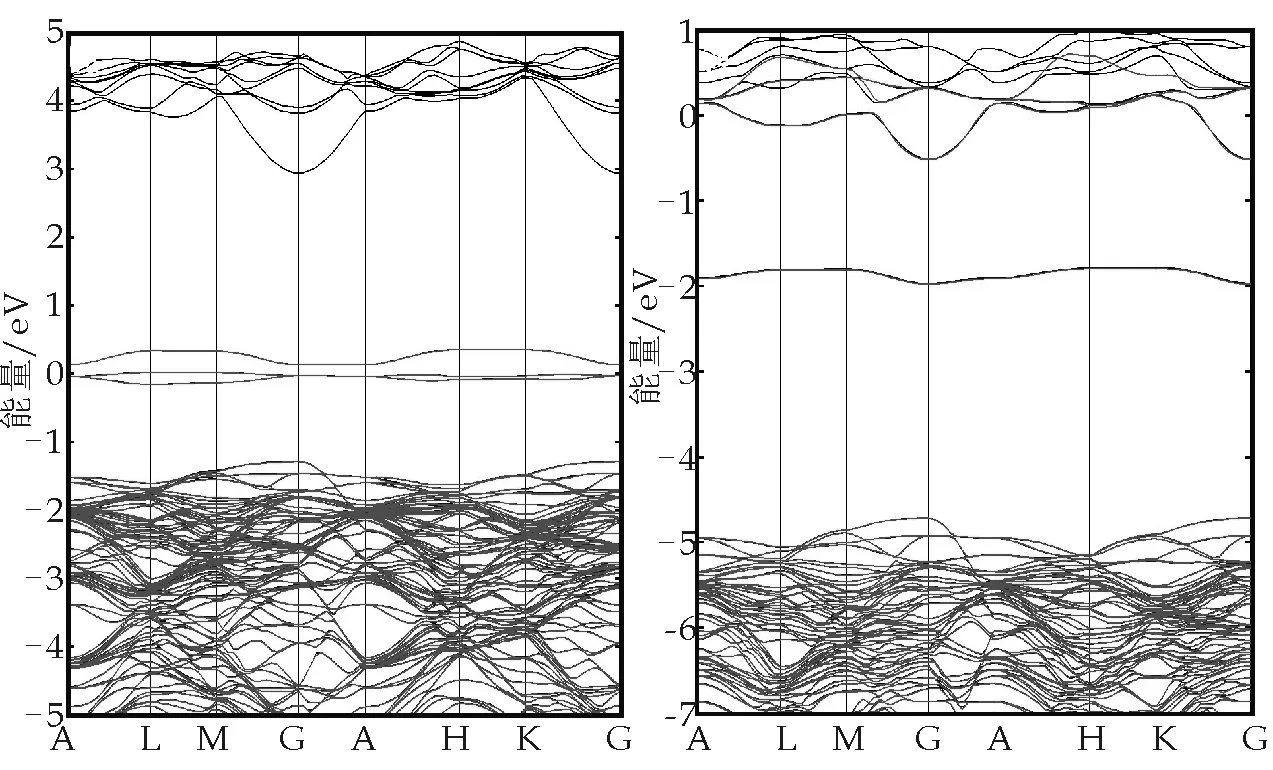
(a)Ni缺陷结构能带 (b)Ali缺陷结构能带结构(黑线)图5 两种间隙缺陷的能带结构,Wannier函数插值得到的能带图用红线表示
2.3.1 Ni电子结构分析
通过观察Ni结构中的Wannier函数中心分布,我们发现在缺陷最近邻以外的结构中,Wannier函数中心的分布已经与理想结构一致,显示Ni对电子结构的影响仅限于最近邻结构之内.
图6(a)给出了缺陷次近邻以内结构中的Wannier函数中心分布,可以看到与间隙最近邻的3个N原子周围的Wannier函数中心位置发生了较大变化,而其余Wannier函数中心位置变化并不明显,这也说明了Ni对晶体电子结构影响较小.缺陷态Wannier函数的形状如图6 (b)所示,为典型的sp3σ成键形状,因此,间隙N原子与其下方距离最近的3个Al原子间成键.

(a)Ni次近邻结构中Wannier函数中心分布,缺陷最近邻Wannier函数用红色小球表示 (b)缺陷近邻处Wannier函数形状图6 Ni结构中Wannier函数形状及中心分布
2.3.2 Ali电子结构分析
相比于Ni,Ali结构优化后近邻原子位置发生了较大变化,显示电子结构变化剧烈.我们首先观察了Ali结构中Wannier函数中心分布,发现次近邻以外的Wannier函数中心位置有了微弱的变化,显示Ali对晶体电子结构的影响在本文研究的四种缺陷结构中最大.
图7(a) 给出了次近邻以内结构中的Wannier函数中心分布,可以看到缺陷周围的最近邻的Wannier函数中心位置变化明显,最下方的Wannier函数中心处在间隙原子与近邻3个Al原子构成的四面体中并靠近间隙原子.由于该Wannier函数对应费米能级下方的电子全部填充的缺陷态能带,因此,它反映了晶体中引入缺陷后孤对电子分别靠近间隙原子及最近邻的N原子.
缺陷上方的3个Wannier函数中心对应费米能级之上的3条能带,由于这几条能带已经进入到导带,其Wannier函数形状变得离散,不再局域,如图7(c)所示.通过观察图7(b)及(c)所给出的Wannier函数形状,尽管Al原子距离上方3个N原子最近,它们之间并没有成键,显示间隙Al原子与周围近邻原子作用较弱,与GaN中的间隙Ga原子表现出的性质一致[6].
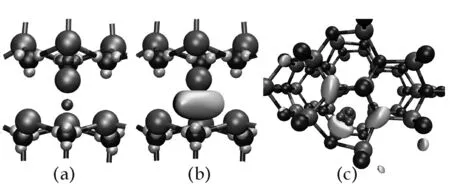
(a)Ali次近邻结构中Wannier函数中心分布,缺陷最近邻Wannier函数用红色小球表示 (b)间隙原子下方Wannier函数形状 (c)间隙原子上方Wannier函数形状图 7 Ali结构中Wannier函数形状及中心分布
3 结论
本文以第一性原理计算为基础,计算了纤锌矿AlN中反位和间隙缺陷结构中的最大局域化Wannier函数,并根据Wannier函数的形状及中心位置分布分析了缺陷引起的电子结构变化及成键信息.研究结果表明,最大局域化Wannier函数方法直观地显现了缺陷结构中的电子结构变化,并给出了清晰的物理图景.
[1] Taniyasu Y,Kasu M,Makimoto T. An aluminium nitride light-emitting diode with a wavelength of 210 nanometres[J].Nature,2006,441(7 091):325-328.
[2] Walker D,Zhang X,Kung P, et al.AlGaN ultraviolet photoconductors grown on sapphire[J].Appl.Phys.Lett.,1996,68(15):2 100-2 101.
[3] Nishida T,Saito H,Kobayashi N.Efficient and high-power AlGaN-based ultraviolet light-emitting diode grown on bulk GaN[J].Appl.Phys.Lett.,2001,79(6):711-712.
[4] Marzari N,Vanderbilt D.Maximally localized generalized Wannier functions for composite energy bands[J].Phys.Rev.B,1997,56(20):12 847-12 865.
[5] Benson D,Sankey O F,Häussermann U.Electronic structure and chemical bonding of the electron-poor II-V semiconductors ZnSb and ZnAs[J].Phys.Rev.B,2011,84(12):125 211-125 219.
[6] Gao F,Bylaska E J,El Azab A, et al.Wannier orbitals and bonding properties of interstitial and antisite defects in GaN[J].Appl.Phys.Lett.,2004,85(23):5 565-5 567.
[7] Corsetti F,Mostofi A A.System-size convergence of point defect properties:The case of the silicon vacancy[J].Phys.Rev.B,2011,84(3):35 209-35 218.
[8] Marzari N,Mostofi A A,Yates J R,et al.Maximally localized Wannier functions:Theory and applications[J].Rev. Mod. Phys.,2012,84(4):1 419-1 475.
[9] Giannozzi P,Baroni S,Bonini N,et al.Qunatum espresso:A modular and open-source software project for quantum simulations of materials[J].J Phys Condens Matter,2009,21(39):395 502-395 521.
[10] Mostofi A A,Yates J R,Lee Y S,et al.Wannier 90:A tool for obtaining maximally-localised Wannier functions[J].Comput.Phys.Commun,2008,178(9):685-699.
[11] Abu Farsakh H,Qteish A.Ionicity scale based on the centers of maximally localized Wannier functions[J].Phys.Rev.B,2007,75(8):85 201-85 207.
[12] Stampfl C,Van de Walle C G.Theoretical investigation of native defects,impurities,and complexes in aluminum nitride[J].Phys. Rev.B,2002,65(15):155 212-155 220.
