化学前沿(2013年10月发布)
2014-03-13汤森路透
汤森路透
化学前沿(2013年10月发布)
汤森路透
“化学前沿”从化学学科的角度报告了生物医药研究领域最新的理念和成就。报告中的信息和观点来源于汤森路透IntegritySM数据库。该数据库结合了生物、化学与药物学等学科的大量信息。本期报告分为有机合成路线展示、研究中的分子骨架、新化合物的作用机制、新研发管线等几个部分,从不同方面描述了对疗效和安全性更好的药物的筛选过程,并介绍了孤儿药的研究现状。
生物医药研究;有机合成;分子骨架;孤儿药
本文介绍了一种新的多肽合成的自然连接方法——丝氨酸/苏氨酸多肽连接(serine/threonine ligation,STL)法,该方法由香港大学李学臣及其团队提出并申请了专利。新的具有生物活性的化学分子骨架结构是推动医药化学学科发展的主要动力,文中列举了8个2013年最新报道的新化学分子骨架。此外,本文还简要介绍了几种新分子作用机制及进入研发视线的新分子实体(new molecular entities,NMEs)。综述了在欧洲、美国、日本将上市的孤儿药,分析了在过去5年中市场上已有的孤儿药,并比较了各种孤儿药的上市数量和专利申请数量,阐述了医药行业是如何逐步认识到孤儿药的研发在投资相对较小的情况下可能会带来更大的经济收益。
1 有机合成路线图展示
蛋白质的合成通常需要两步。第一步,通过固相多肽合成技术(solid-phase peptide synthesis,SPPS)产生中等长度的多肽片段;第二步,通过化学连接,将这些片段选择性地拼接在一起。
有效的片段连接方法是多肽缩合成功的关键。由Kent等人在20世纪90年代中期提出的自然化学连接(native chemical ligation,NCL)是这一领域的转折点[1]。在此方法中,一个C端为硫酯的未保护多肽片段和另一个N端为半胱氨酸残基的未保护多肽片段进行反应,在结合位点处形成天然肽键。但是该方法需要多肽中含有半胱氨酸残基,而半胱氨酸在天然蛋白质中含量相对较低,于是研究者们开始寻找不依赖巯基的连接方法,其中许多都要加入烦琐的步骤使反应具有选择性。
香港大学李学臣及其团队提出在丝氨酸/苏氨酸位点进行连接:连接C端邻醛基酚酯和N端丝氨酸或苏氨酸1,2-羟胺部分,产生N,O-苯亚甲基缩醛中间体,从而方便地获得具有天然连接位点的连接产物。该策略已初步用于一个模型二肽的合成[2],但尚未应用于真正意义上的蛋白质合成。
目前,研究者有充分证据表明此方法可与标准SPPS兼容,并可在没有保护基团的情况下实现连接,这一点与NCL相似。
这个团队已将此方法成功用于合成相对分子质量为11 000、由98个氨基酸残基组成的蛋白质——酰基磷酸酯酶,一种具有重大生物学价值的人红细胞酶[3]。李学臣等人的成功表明此方法在蛋白质合成中展现出良好的发展前景,原因是大量的天然蛋白质中都有许多丝氨酸和苏氨酸残基,尤其是经过翻译后修饰的蛋白质(如磷酸化或糖基化的蛋白质)。
抗癌疫苗的开发是利用合成蛋白质前景最看好的领域之一。富含丝氨酸和苏氨酸的大型MUC1糖蛋白在乳腺、前列腺、卵巢和直肠恶性上皮细胞中表达,并与癌症的发展和转移息息相关。目前已生产出几种MUC1糖多肽,包括VXL-100( 数据库条目编号:695747) 和emepepimut-S( 数据库条目编号:243623),作为保护剂来对抗这种活性。李学臣的团队目前已将STL 法应用于蛋白质合成,如通过3 次迭代连接生产出含有8 个多糖的80 聚体MUC1 片段[4]。
该团队也将注意力转向达托霉素(1)[5]。达托霉素( 数据库条目编号:111916) 是由礼来公司的科学家从土壤微生物Streptomyces roseoporus 中分离出的一种环状脂质缩酚酸肽。该化合物可用于治疗由耐甲氧西林金葡菌(MRSA)等引起的革兰阳性皮肤感染,且尚未对细菌产生耐药性。该团队已使用其专利保护的STL 法首次实现了这种抗生素的全合成。该混合固/ 液相方案可替代基因工程来开发新的类似化合物[6]。
达托霉素的成功取决于STL 法也可用于分子内,如图1 所示,在同一个多肽中C 端为邻醛基酚酯的侧链未保护多肽可与N 端丝氨酸或苏氨酸反应。中间体可形成一个流畅的大环内酰胺反应,使纯化后的达托霉素产率达到67%。

图1 达托霉素的合成路线Figure 1 Synthesis route of daptomycin
采用STL 法合成已知的蛋白质及其类似物取得的各种成功展现了此方法在多肽连接中的应用前景,并为用于治疗从细菌感染到癌症的复杂分子的合成以及这些蛋白质的生物学意义的阐释提供了新的可能性。
2 研究中的分子骨架
此节中所展示的一系列新的分子骨架代表着最新的合成或由天然产物衍生出的结构,该类结构可为今后的新药研发奠定基础。
2.1 抗疟药物
华东理工大学的团队希望通过寻找plasmepsin Ⅱ抑制剂来发现新的抗疟药物[7]。Plasmepsin Ⅱ被研究者认为是“最具吸引力的抗疟药物靶标”。他们对2 种化学数据库进行了基于结构的虚拟筛选,成功地鉴定出此蛋白的抑制剂,确认了5 个具有新型非肽类分子骨架的苗头化合物(代表化合物:2,数据库条目编号:812359),在低于10 μmol·L-1 时具有中等抑制活性。如每个分子的对接模拟实验显示,这些抑制剂与蛋白质通过多种氢键作用稳定结合。
2.2 耐药性癌症候选治疗药物

日本自治医科大学和兴和株式会社的研究人员致力于蛋白酶体抑制剂的研究,希望从中发现新的治疗耐药性癌症的方法[8]。蛋白酶体通过分解多泛素化修饰后的蛋白质维持细胞稳态。这类抑制剂可以成为有效的癌细胞杀手,事实上,硼替佐米就是这样一种抑制剂,它在治疗多发性骨髓瘤中不可或缺。可惜的是,该药只能静脉注射,并且有耐药性。该团队将高哌嗪衍生物(HPDS)看做是具有良好前景的候选药物,已经证明其对从各种血液系统恶性肿瘤(包括多发性骨髓瘤)的细胞系具有细胞毒性。此外,其中的一个化合物K-7174(3,数据库条目编号:291665),能够抑制耐硼替佐米的骨髓瘤细胞的生长,并与此药具有叠加效应。

2.3 黑色素瘤治疗药物
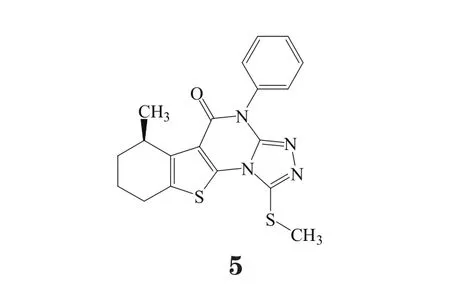
以丝氨酸/ 苏氨酸蛋白激酶B-raf(B-Raf 激酶)抑制剂作为溶瘤药物是来自中国南京大学、中国科学院和中南大学以及Wistar 研究所的研究人员关注的焦点[9]。他们解释了8% 的恶性肿瘤病例和50% 的黑色素瘤病例存在V600E BRAF 激酶突变的原因。该突变可激活下游MAPK 信号通路。该团队已经用虚拟筛选和化学合成方法确定了一系列的N-( 噻吩-2 ) 苯甲酰胺衍生物,它们是潜在的强效BRAF(V600E)抑制剂。对接研究表明,其中一个先导化合物(4,数据库条目编号:813320)通过独特的结合模式(包含倒置方向)与BRAF(V600E)的活性位点结合发挥疗效。

2.4 抗炎药物
趋化因子CXCR2(IL-8Rβ)受体拮抗剂被美国南加州大学(USC)研究人员证实可成为潜在的抗炎药物[10]。 CXCR2 由免疫细胞表达,在感染或损伤后募集中性粒细胞,并启动血管生成。在过去的几十年里,CXCR2 抑制剂未能成功进入临床阶段。该小组目前采用一种高通量的、基于细胞的CXCR2 检测方法在自建的化合物数据库中进行筛选,CX-797(5,数据库条目编号:813976)被确定为先导化合物。
研究小组认为CXC-797 能抑制IL-8 介导的环磷酸腺苷信号传导和受体的降解,同时能上调IL-8 介导的β-arrestin 的募集和MAP 激酶信号传导。这种偏向性的CXCR2 配体信号传导表明,与之前报道的CXCR2拮抗剂相比,该化合物具有独特的作用方式,因此对一系列炎症性疾病和癌症的治疗具有潜在的优化和开发前景。
2.5 治疗癌症的活性化合物
华东理工大学的科研人员研究了有望成为溶瘤细胞药物的法尼基转移酶(FTase,也被称为牛儿基焦磷酸合成酶)抑制剂的作用模式[11]。他们将基于结构的虚拟筛选用于发现微摩尔范围内对MCF7 细胞有中等抑制活性的化合物。筛选出的22 个苗头化合物(代表化合物:6,数据库条目编号:820381),可用18 个不同的分子骨架代表作为后续苗头化合物- 先导化合物优化的新型化学类型。
2.6 TRPM8 受体拮抗剂
日本静冈县立大学、城西大学和千叶大学的科学家们在研究可作为TRPM8 (TRP-p8; CMR1) 受体拮抗剂的伏康京碱(7,数据库条目编号:401403)[12]。TRPM8可被低温和具有降温作用的化合物如薄荷醇激活,对冷痛觉异常和泌尿系统、呼吸系统疼痛具有重要作用,因此寻找这一领域的新型抑制剂将成为解决上述问题的方法。可惜的是,人类对天然拮抗剂尚知之甚少。该团队目前在Voacanga africana 树上发现了一个具有iboga 骨架的有效的TRPM8 受体拮抗剂伏康京碱。该化合物可在比辣椒平或其他天然抑制剂浓度低得多的情况下减弱对由薄荷醇或icilin 诱导的TRPM8 的激活,但其效果略弱于众所周知的拮抗剂N-(4- 叔丁基苯基)-4-(3- 氯吡啶-2- 基) 哌嗪-1- 甲酰胺(BCTC)。该研究小组认为,大多数熟知的TRPM8 受体拮抗剂都是非竞争性的,而作为竞争性抑制剂的伏康京碱很可能会成为一类全新的TRPM8 受体阻滞剂。

2.7 天然来源的泛素羧基末端水解酶抑制剂
Spongiacidi C(8,数据库条目编号:820456)是一直在寻找可作为溶瘤细胞药物的泛素羧基末端水解酶7(USP7) 抑制剂的美国Progenra 公司科学家们关注的焦点[13]。USP7 是可作为能水解泛素C 端异肽键的脱泛素酶的一个新兴的癌症靶标。Progenra 团队从海绵Stylissa massa 中分离出spongiacidin C,成为第一个天然来源的USP7 抑制剂。其IC50 为3.8μmol·L-1,在该类抑制剂中测试结果最佳。过去是通过筛选小分子合成数据库去发现USP7 抑制剂,而来自海洋无脊椎动物的结构丰富、具有潜在生物活性的天然产物为我们提供了一个有效的先导化合物。

2.8 镇痛药
Esteve 的科学家致力于采用高通量筛选的方法寻找非阿片类止痛药,发现了一系列新的六氢-2H- 吡喃并[3,2-c] 喹啉衍生物[14]。他们已经合成了这些化合物并研究了其药理活性。这些化合物均为强效的σ1 受体配体。重要的是,它们不像其它受体配体那样含有强碱性氨基,而是含有一个类似苯胺的弱碱性氮,且这些化合物对σ2 受体具有选择性。该团队的数据库中的一个化合物具有亚微摩尔级亲和力,并且该团队在苗头化合物- 先导化合物研究项目中发现了一个活性显著提高、理化性质有所改善的新化合物(9,数据库条目编号:814387),该化合物对辣椒素和福尔马林模型中的小鼠神经性疼痛具有镇痛作用。

3 新分子作用机制
一个化合物的作用机制对于提高分子设计水平以及发现更加有效且副作用较小的先导化合物非常关键。此部分列出了一些热点化合物(见表1)。

表1 具有新作用机制的热点化合物Table 1 A range of facinating compounds with new molecular mechanisms of action

4 新研发管线
新研发管线指刚刚进入研发视线的NMEs。本节将介绍丙型肝炎、癌症、糖尿病以及其它一些病症如疟疾、精神分裂症和骨关节炎相关的药物( 见表2)。

表2 进入研发领域的新分子实体Table 2 New molecular entities ready to progress into the R&D arena

续表2

5 孤儿药的研发现状
药物研发让我们有了新的认识:我们可以活得更长久并且更健康。因为药物可以快速有效地治疗感染,解决常见的疾病症状,明显提高癌症患者的存活率。此外,药物还能治疗由老龄化引起的退行性疾病。但是,世界上仍然有许多罕见的、特殊的、目前还没有针对性治疗药物的疾病。2002年美国《罕见病法案》将罕见病或“孤儿病”定义为发生率低于1/1 500的疾病,欧洲相关组织则将疾病对生命的威胁程度而非发生率作为决定性因素。
有些罕见病全球只有十几个患者,但发生率为1/1 500的疾病则能影响到数百万人。据估计,美国有2 500万罕见病患者,欧洲约有3 000万。罕见病种类繁多,约7 000种,且随着250种新病症的出现,这个数目每年都在增加。新增的罕见病包括阿斯科格综合征、戈谢病、Hunter综合征、1型酪氨酸血症、卡勒病、瓦登伯格无眼畸形综合征和接合菌病等。
与罕见病对应的治疗药物便是孤儿药,即可以用来治疗罕见病的药物。由于这些药物能够满足之前尚未得到满足的医疗需求,因此在国际上往往能够较快通过药物审核。而且,为了补偿用药人群相对较少造成的损失,行业可能会对审核中的药品索价较高。
基本上,孤儿药能够帮助患者改善身体状况;而对于医药行业本身而言,孤儿药是一种至今未被开发的替代药物,它所能带来的收入可能会抵消所谓“重磅炸弹”药物的消亡,使逐渐干涸的药物开发河流重新流淌,使之前获得巨大利润而专利到期的药物复活[31]。
医药行业一直在进行罕见疾病的研究和相应药物的开发。自欧盟2000年《孤儿药条例》通过以来,已有1 000余种药物被列为孤儿药[32],1983年美国《孤儿药法案》确立以来,已有近3 000种药物被列为孤儿药[33],而日本1993年通过《药事法规》后,20年来已有300多种药物被列为孤儿药[34]。虽然对于同一疾病,有些新增药物与之前药物相同,但对于治疗一些非常罕见却严重的疾病,这些药物还是有非常显著的进步。
然而,一份关于近5年孤儿药退出药品市场的报告分析表明,孤儿药并非一定能够产生良好的预后效果。无论是NCEs还是线性延伸的药物,2009年以孤儿药身份首次上市的仅有6种,2010年、2011年和2012年也分别仅有7、11和12种。据估计,2013年新上市孤儿药可能有17种,是2009年的近3倍,这个数字包括了潜在的可能上市的药物,所以接下来几个月并不会再增加。
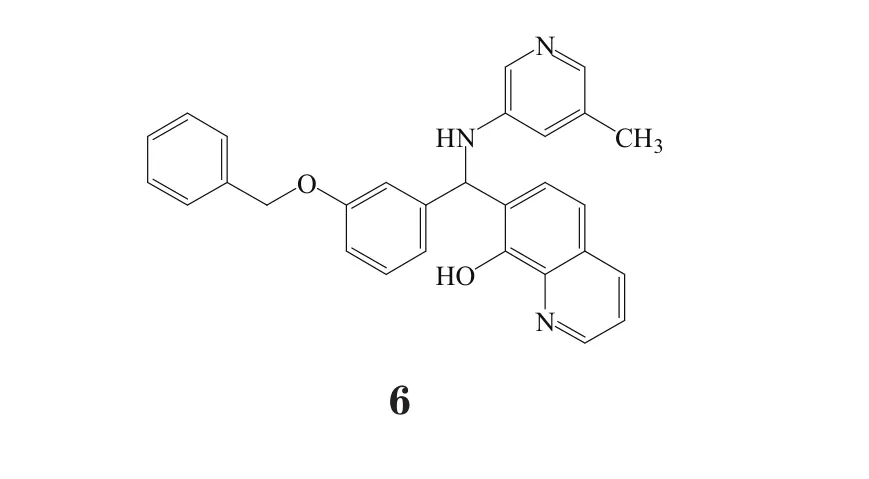
值得关注的是,类似于lomitapide(数据库条目编号:235523,商品名:Juxtapid™,Aegerion制药公司)和米泊美生钠(数据库条目编号:339511,商品名:Kynamro™,健赞公司罕见病研究中心)的药物,这2种药物都可以用于治疗家族性高胆固醇血症,是罕见病治疗药物的里程碑。Lomitapide也是一种一线治疗药物,该药具有全新作用机制,作为微粒体三酰甘油酯转运蛋白抑制剂上市。同样还有其他3种药物:用于治疗短肠综合征的teduglutide(数据库条目编号:246346,商品名Gattex®,NPX制药公司)、2013年通过审核的用于治疗黑色素瘤的药物甲磺酸达拉非尼(数据库条目编号:679978,商品名Tafinlar®,葛兰素史克公司)和二甲基亚砜曲美替尼(数据库条目编号:415494,商品名Mekinist™,葛兰素史克公司)。
治疗某一罕见病的药物专利数目每年都会发生变化,比如治疗囊性纤维化的专利药物,2009年有68个,而2010年上升到了80个左右(见图2);治疗多发性骨髓瘤的药物数目也发生了相似的变化(见图3);对于另一个更为罕见的疾病——2型黏多糖贮积症(Hunter综合征),自2009年以来,共有11个专利药物(见图4)。

图2 近几年用于治疗囊性纤维化的药物专利数变化Figure 2 Number of patent families filed by year,related to cystic fibrosis disease
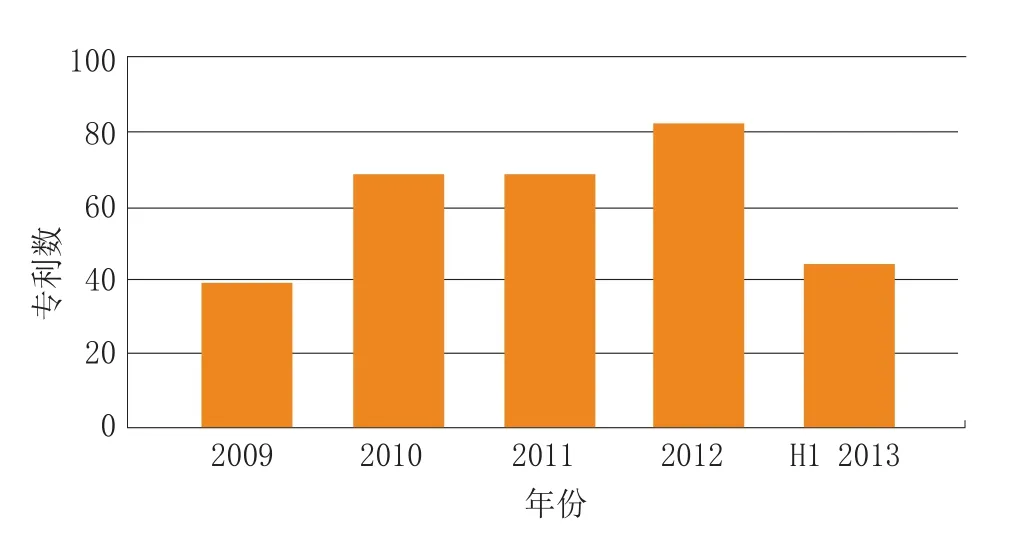
图3 近几年用于治疗多发性骨髓瘤的药物专利数变化Figure 3 Number of patent families filed by year,related to multiple myeloma disease

图4 近几年用于治疗Hunter综合征的药物专利数变化Figure 4 Number of patent families filed by year,related to MPS type II Hunter disease
随着现代医学诊断技术的发展,罕见病的种类会越来越多,罕见病患者数目也会越来越多。制药行业一直致力于寻找“重磅炸弹”药物或者能带来巨额利润的市场规模巨大的药物。而现在,他们也逐渐意识到孤儿药具有低投入、高回报的发展潜力。能够快速通过审核程序、临床试验规模较小等能够大大降低这类药物的研发费用。
解决罕见病问题对于全世界的医药领域都迫在眉睫。全球领先的制药公司,包括上文提到的健赞、葛兰素史克,以及Shire、辉瑞、诺华、Prosensa和BioMarin等公司,都已经与美国罕见病组织以及欧洲罕见病组织联合,以期解决罕见病问题。
[1]Dawson P E,Muir T W,Clark-Lewis I,et al. Synthesis of proteins by native chemical ligation[J]. Science,1994, 266(5186): 776-779.
[2]Li X,Lam H Y, Zhang Y, et al.Salicylaldehyde ester-induced chemoselective peptide ligations: enabling generation of natural peptidic linkages at the serine/threonine sites[J]. Org Lett, 2010, 12(8): 1724-1727.
[3]Zhang Y, Xu C, Lam H Y,et al. Protein chemical synthesis by serine and threonine ligation[J].Proc Natl Acad Sci USA,2013, 110(17): 6657-6662.
[4]Xu C, Lam H Y, Zhang Y, et al. Convergent synthesis of MUC1 glycopeptides via serine ligation[J]. Chem Comm,2013, 49(55): 6200-6202.
[5]Lam H Y, Zhang Y, Liu H,et al. Total synthesis of daptomycin by cyclization via a chemoselective serine ligation[J].J Am Chem Soc, 2013, 135(16): 6272-6279.
[6]Li X. Native chemical ligation at serine and threonine sites: WO 2011017837[P]. 2011-02-17.
[7]Song Y,Jin H,Liu X,et al. Discovery of non-peptide inhibitors of Plasmepsin II by structure-based virtual screening[J]. Bioorg Med Chem Lett,2013, 23(7): 2078.
[8]Kikuchi J,Shibayama N, Yamada S,et al. Homopiperazine derivatives as a novel class of proteasome inhibitors with a unique mode of proteasome binding[J].PLoS ONE,2013, 8(4): e60649.
[9]Xie Y,Chen X,Qin J,et al. Identifcation and synthesis of N-(thiophen-2-yl) benzamide derivatives as BRAF(V600E) inhibitors[J].Bioorg Med Chem Lett,2013, 23(8): 2306.
[10]Ha H,Neamati N. Discovery and mechanistic studies of novel class of CXCR2 inhibitors. 104th Annu Meet Am Assoc Cancer Res (AACR),Washington, D.C ,April 6-10[C]. 2013, Abst 4558.
[11]Yu X J,Zhao X,Zhu L L,et al. Discovery of novel inhibitors for human farnesyltransferase (hFTase) via structure-based virtual screening[J]. Med Chem Commun,2013, 4(6): 962.
[12]Terada Y,Horie S,Takayama H, et al. New class TRPM8 antagonist, voacangine is the first natural competitive antagonist for menthol receptor, TRPM8. 245th ACS Natl Meet,New Orleans,April 7-11[C]. 2013, Abst MEDI 384.
[13]Yamaguchi M,Miyazaki M,Kodrasov M P,et al. Spongiacidin C, a pyrrole alkaloid from the marine sponge Stylissa massa, functions as a USP7 inhibitor[J].Bioorg Med Chem Lett,2013, 23(13): 3884.
[14]Diaz J L,Christmann U, Fernandez A, et al. Synthesis and biological evaluation of a new series of hexahydro-2H-pyrano(3,2-c)quinolines as novel selective delta1 receptor ligands[J]. J Med Chem,2013,56(9): 3656.
[15]Murugesan D,Mital A,Kaiser M,et al.Discovery and structure-activityrelationships of pyrrolone antimalarials[J].J Med Chem,2013,56(7):2975.
[16]Gu Z, Graci J D,Lahser F C,et al. Identifcation of PTC725: An orally bioavailable small molecule that selectively targets the hepatitis C virus NS4B protein[J]. Antimicrob Agents Chemother, 2013, 57(7): 3250.
[17]Graci J D,Gu Z,Jung S P,et al. Identification of PTC725: A potent, selective and orally bioavailable small molecule that targets the hepatitis C virus NS4B protein. 26th Int Conf Antivir Res (ICAR), San Francisco, May 11-15[C]. 2013,Abst 18.
[18]Karp G M,Zhang X,Zhang N,et al. Chemical optimization of a novel class of 6-(indol-2-yl)pyridine-3-sulfonamides targeting HCV NS4B: Potent and orally bioavailable compounds with an improved ADMET profile. 26th Int Conf Antivir Res (ICAR), San Francisco, May 11-15[C].2013,Abst 53.
[19]Karp G M,Zhang X,Zhang N, et al. Discovery and SAR optimization of N-(hetero)aryl-6-(indol-2-yl)pyridine-3-sulfonamides: PTC725, a potent, selective and orally bioavailable development candidate targeting HCV NS4B. 26th Int Conf Antivir Res (ICAR), San Francisco, May 11-15[C]. 2013,Abst 52.
[20]Yano F,Hojo H,Ohba S,et al. A novel disease-modifying osteoarthritis drug candidate targeting Runx1[J]. Ann Rheum Dis,2013, 72(5):748.
[21]Yano F,Hojo H,Ohba S,et al. Cell-sheet technology combined with a thienoindazole derivative small compound TD-198946 for cartilage regeneration[J].Biomaterials,2013,34(22):5581.
[22]Carpenter K J,Brog R,Moreno C,et al.Small molecule inhibitors of PIM kinases as potential treatments for urothelial carcinomas. 104th Annu Meet Am Assoc Cancer Res(AACR), Washington, D.C.,April 6-10[C]. 2013,Abst 2174.
[23]Maciag A E, Saavedra J E,Holland R J,et al.GSTP-activated nitric oxide-releasing/PARP inhibitor hybrid prodrugs induce cancer cell death through ROS/RNS, DNA damage, ER stress, and apoptosis. 104th Annu Meet Am Assoc Cancer Res (AACR), Washington, D.C. ,April 6-10[C]. 2013,Abst 3334.
[24]Revel F G,Moreau J L,Pouzet B,et al. A new perspective for schizophrenia: TAAR1 agonists reveal antipsychotic- and antidepressantlike activity, improve cognition and control body weight[J]. Mol Psychiatry ,2013,18(5):543. Norcross R D,Galley G,Zbinden K G,et al. Discovery and
[25]characterisation of (S)-4-((S)-2-phenyl-butyl)-4,5-dihydro-oxazol-2-ylamine (RO5256390) - A TAAR1 agonist clinical candidate for treatment of schizophrenia.Annu Meet Frontiers Med Chem, San Francisco,June 23-26[C]. 2013,Abst 22. Huang H,Acquaviva L, Bregman H,et al.Structure-based design
[26]of aminopyridine oxazolidinones as potent and selective tankyrase inhibitors.Annu Meet Frontiers Med Chem, San Francisco, June 23-26[C]. 2013,Abst 71. Hu L,Magesh S,Chen L,et al.Discovery of a small-molecule inhibitor
[27]and cellular probe of Keap1-Nrf2 protein-protein interaction[J].Bioorg Med Chem Lett,2013,23(10):3039. Fitzgerald K,Bergeron M, Willits C, et al. Pharmacological inhibition
[28]of Polo Like Kinase 2 (PLK2) does not cause chromosomal damage or result in the formation of micronuclei[J].Toxicol Appl Pharmacol,2013, 269(1): 1. Bowers S,Truong A P,Ye M,et al. Design and synthesis of highly
[29]selective, orally active Polo-like kinase-2 (Plk-2) inhibitors[J].Bioorg Med Chem Lett,2013,23(9):2743. Goto T,Nagamine J,Hiramine Y,et al. Effect of DSP-0011, a novel
[30]selective 11beta-hydroxysteroid dehydrogenase type 1 inhibitor,on visceral adiposity in diet-induced-obese mice and common marmosets. 95th Annu Meet Endo Soc, San Francisco, June 15-18[C]. 2013,Abst FP03-3. Sharma A, Jacob A, Tandon M, et al. Orphan drug: development trends
[31]and strategies[J]. J Pharm Bioallied Sci, 2010, 2(4): 290-299. European Commission. 1,000th designation of an orphan medicinal
[32]product: improving the lives of patients [EB/OL]. (2012-06-07) [2013-10-14]. http://europa.eu/rapid/press-release_IP-12-573_ en.htm?locale=en.
[33]U.S.Food and Drug Administration. Search Orphan Drug Designations and Approvals. [EB/OL]. [2013-10-14]. http://www.accessdata.fda.gov/ scripts/opdlisting/oopd/index.cfm.
[34][Anon].List of products designated as orphan drugs for rare diseases in Japan[EB/OL]. [2013-10-14]. http://www.nibio.go.jp/shinko/orphan/ english/pdf/h2507kisyoiyaku-hyo1.pdf.
编者注:本文来源于汤森路透“制药事务报告”
(2013 年 10 月发布),内容有删改,格式经调整。
The Cutting Edge of Chemistry(Issued October 2013)
Thomson Reuters
The Cutting Edge of Chemistry discloses new ideas and achievements in the biomedical research feld with the chemist’s perspective in mind.Insights and information within this report are drawn from Thomson Reuters IntegritySM,the drug discovery database that combines biology,chemistry and pharmacology information to empower your research.The report has been organized into sections, including organic synthesis scheme showcase,scaffolds on the move,new molecular mechanisms of action and the starting line that delineate essential aspects of the search for better and safer drugs.And the research status of orphan drugs has also been introduced.
biomedical research;organic synthesis;scaffold;orphan drug
R392; R593.2
A
1001-5094(2014)12-0949-09
