农作物病原卵菌基因组数据库资源概述
2013-09-11高亚梅王相晶向文胜
高亚梅, 王相晶, 向文胜*
(1.东北农业大学生命科学学院,哈尔滨 150030;2.黑龙江八一农垦大学生命科学技术学院,大庆 163319)
卵菌(Oomycetes)属于色藻界(Chromista),包括腐生菌及植物、动物和许多微生物的病原菌。病原卵菌导致许多作物、花卉等发生灾难性病害,例如引起大豆疫霉根腐病的大豆疫霉(Phytophthora sojae Kaufmann et Gerdemann);引起的马铃薯晚疫病的 致 病 疫 霉 [P.infestans (Montagne)de Bary][1-2]。在进化地位上,卵菌与褐藻和硅藻有较近的亲缘关系,形成一个独特的二倍体微生物类群[3],其分子遗传学的研究相对落后。随着分子生物学技术的发展,卵菌基因组学及功能基因组学的研究逐渐受到研究者的重视,迅速积累了大量基因组数据资源,这些资源又成为卵菌功能基因组学研究的基础之一,同时也是基因组数据挖掘的重要来源,加快了卵菌分子生物学的各项研究。本文对农作物病原卵菌基因组数据库资源及其检索方法做一概述。
1 卵菌结构基因组学数据库资源及检索方法
随着测序技术的不断发展,高通量测序使得测序成本不断降低,越来越多的基因组计划启动,甚至可以用宏基因组学(metagenomics)技术测定一个生物群体的序列。因此,在各类数据库中积累了海量的生物序列信息,供研究者检索、下载和分析。基因组序列及结构、基因功能和调控元件的相关注释可为深入研究病原菌毒力、致病性、寄主特异性调控、病原菌生化与生理机制、生态位适应性等提供大量有用信息。目前,共有7个卵菌物种[大豆疫霉P.sojae、分枝疫霉P.ramorumWerres,De Cock et Man、致病疫霉 P.infestans、辣椒疫霉P.capsici Leonian、终极腐霉 Pythium ultimum Trow、Hyaloperonospora parasitica (Gäum.)Göker,Riethm.,Voglmayr,Weiss & Oberw.和古巴假霜霉 Pseudoperonospora cubensis (Berkeley et Curtis)Rostovzev]的全基因组序列正式对外公布(详见表1)[4-9];另外2个卵菌物种(Peronospora parasitica Tul.及Saprolegnia parasitica Coker)的基因组测序工作正在进行中。通过对这些基因组序列和结构的分析揭示了卵菌基因组在大小、致病相关基因、专性活体寄生等方面存在多样性[10],并为病原菌遗传与致病机理、宿主-病原菌相互作用的特征(宿主的特异性,毒性策略等)以及宿主-病原菌相互作用的进化等各方面研究提供了大量的信息。基因组分析揭示的卵菌基因组进化特征包括重复序列在基因组内的扩增、缺失、基因融合和基因垂直转移等[11-13]。这些无疑都加深了我们对于卵菌基因组结构及进化的认识。

表1 已公布的测序卵菌基因组信息资源Table 1 Oomycete sequenced genome database resources
联合基因组研究所JGI网站(DOE Joint Genome Institute)提供细菌、古细菌、真核生物和宏基因组等众多物种的基因组序列信息和分析服务,在其主页的搜索工具栏选择相应的物种即可进入相应物种的数据库,其中卵菌物种包括P.sojae、P.ramorum和P.cinnamomi[14]。基因组序列信息可通过该数据库SEARCH功能进行关键词检索,利用BLAST进行序列相似性搜索,利用DOWNLOAD下载,利用BROWSE以图形化方式浏览(见图1)。另外 GO、KEGG、KOG、CLUSTER、SYNTENY提供基因功能、代谢图谱、共线性、簇等注释信息的查询检索。

图1 JGI网站P.cinnamomi var.cinnamomi基因组浏览界面Fig.1 The genome browse interface of P.cinnamomi var.cinnamomi in JGI
VMD数据库(VBI Microbial Database)[15]是另一重要的卵菌基因组数据库,包括Pythium ultimum(V1.0),Phytophthora infestans(V4.0),P.capsici(V11.0),P.sojae(V1.0,V4.0,V5.0),P.ramorum(V1.0),H.arabidopsidis(V3.0,V6.0,V8.3)基因组及注释信息。每个数据库均提供图形化的浏览方式,显示每个基因的位置、模式和相关基因组的BLAST信息等。VMD数据库还提供检索、下载、注释和工具箱等服务。检索可以通过不同方式进行,包括 Gene ID、GO ID、Scaffold Number、protein domains/motifs/functions、primary annotation、affymetrixID 等。检索结果页面提供基因的序列、基因组位置、功能注释及软件预测结果等信息。BLAST工具提供多个可选数据库,包括已测序卵菌基因组、表达数据库、大豆和拟南芥数据库等,进行有针对性的序列相似性比对分析。P.infestans T30-4基因组序列可以在Phytophthora infestans Database数据库下载,该数据库同样提供各类分析工具和可视化的浏览页面。
Pythium Genome Database是专门存储Pythium ultimum序列信息的数据库,包括其代表性菌株Pythium ultimum DAOM BR144的基因组序列和不同营养条件下的菌丝体的EST序列。在Pythium Genome Database可以下载 Genome Assembly、Transcripts、Proteins和Gene Model GFF3的信息。通过Genome Browser可视化浏览基因组,可以使用序列名、基因名、遗传位点或区域标记进行检索。BLAST工具可以选择卵菌基因组、表达数据等12个数据库进行序列比对。该数据库还将储存Pythium ultimum另一菌株高通量测序结果,开展对两菌株及它们与其他已测序卵菌的基因组比较研究。
卵菌基因组测序多采用第二代测序技术,随着测序成本的下降,将会有更多的卵菌基因组数据产生,包括致病菌和非致病菌,为功能基因组学和比较基因组学研究提供更多的数据。
2 卵菌功能基因组学数据库资源及检索方法
目前,在各种数据库中均存储了大量的各种类型、代表不同信息的数据,如何对这些数据进行分析,获得蕴含其中的生物学意义是当前生物信息学的重要研究领域。利用生物信息学方法对卵菌基因组内大量未知功能基因的分析比较是基因功能研究和数据注释的重要手段。以下介绍的一些常用的卵菌EST数据库、综合性功能基因组资源平台和专门数据库是开展卵菌功能基因组学研究的重要资源。
2.1 卵菌EST数据库资源
卵菌表达序列标签EST序列(Expressed Sequence Tags,ESTs)的大量公布为进行全面的基因结构和功能研究提供了重要的数据,必将促进对卵菌致病分子机制的认识,从而为防治卵菌病害设计合理有效的策略。目前数据库中的EST序列集中在已正式公布的基因组序列的卵菌物种中,例如已有大豆疫霉(P.sojae)的 EST约33350条,而致病疫霉(P.infestans)有99320条(http:∥phytophthora.vbi.vt.edu/EST),还包括一些正在或已完成测序的卵菌物种P.capsici、Phytophthora brassicae De Cock et al、Albugo candida (Pers.)Roussel等[16]。这些 EST序列大部分为病原卵菌侵染寄主植物过程中的转录组,代表疫霉侵染与致病过程中表达的基因,还包括卵菌不同发育阶段及不同环境条件下的EST。卵菌EST存放于多个数据库中,如基因组的数据库、NCBI、转录组数据库OTD等,提供多种途径可以获取和检索。
OTD卵菌转录组学数据库(Oomycetes Transcriptomics Database,V4.0,http:∥vmd.vbi.vt.edu/transcripts/)是一个整合的转录组和EST数据资源的数据库[17],储存卵菌侵染寄主和不同生长条件下的转录组数据,包括EST序列、NGS(next generation sequencing)转录组序列、数字基因表达谱序列 (Digital Gene Expression Profile,DGE)等。OTD浏览器以可视化方式显示转录本的组装,转录本在基因组上的定位、表达谱及转录本在基因组特定位置覆盖度等信息。该数据库提供多种检索方式,可利用EST序列和重叠群ID号、关键词、表达量等进行检索。来自H.arabidopsidis的37492 ESTs通过聚类形成Unigenes,并利用BLAST、InterProScan、TMHMM、TargetP和SignalP进行了注释。EST序列可从该数据库下载。OTD提供与VMD、PTD数据库的链接界面。疫霉转录组学数据库(Phytophthora transcriptomics database,PTD http:∥phy.njau.edu.cn/ptd)提供大豆疫霉的数字基因表达谱信息[18],可通过基因ID、关键词进行检索,利用序列比对BLAST搜索同源基因,可查看不同发育阶段的差异表达基因。基于转录信息的基因研究是功能基因组学研究的重要策略,例如EST数据不仅为基因的功能研究提供有价值的表达信息,而且基于EST的电子基因克隆技术也可用来发现新基因。近年来,二代测序技术产生的转录组序列为基因功能研究提供了更为有利的工具,例如RNA-Seq产生的转录本信息可以更为有效地发现未知转录本和稀有转录本,提供转录本的结构和表达水平信息,用于精确地识别可变剪切位点以及cSNP(编码序列单核苷酸多态性)[19]。
2.2 卵菌综合性功能基因组资源
综合性疫霉病原菌基因组学资源(Comprehensive Phytopathogen Genomics Resource,CPGR)包括4个数据库:基因组仓库、注释数据库、转录物组装和rDNA数据库[20]。基因组仓库储存包括植物病原菌基因组及EST测序计划信息,每年至少更新2次。注释数据库包含基因组注释信息,包括物种名、所属类群、引起的疾病、NCBI序列登录号、基因组大小、测序计划状态及机构、特征、相关注释以及文献信息等。数据库页面包含依据分类单元和测序状态的过滤器功能。转录物组装数据来自NCBI数据库的EST和mRNA,去除低重复和污染序列后,经拼接注释而成。CPGR的转录物组装和来自GenBank的序列可在其FTP下载。rDNA数据库包括植物病原菌的名称和分类单元标识符,另外该数据库在病原菌和植物病害之间做了超链接。CPGR的搜索工具允许用户使用序列和功能注释搜索该数据库。卵菌注释可以通过位点标识符、功能、Pfam功能域、Interpro功能域搜索。对于已测序的卵菌,用户可在CPGR网站利用其提供的一系列界面实现对基因组信息(序列和注释信息)的浏览、下载和检索。目前只有P.infestans T30-4和Pythium ultimum DAOM BR144可利 用 CPGR 的 genome browser进行可视化浏览,包括等位基因、基因模式(gene models)、rRNA 和tRNA 基因、推测的简单重复序列SSRs(putative simple sequence repeats)、GC 含 量 (GC content)和 六 框 翻 译 (sixframe translation)。位点信息和基因模式与其他内容形成超链接。基因组浏览器还包括基因列表(gene list)、RNA 基因列表(RNA gene list)、Pfam功能域列表、Interpro功能域列表、推测SSRs,缩放工具允许用户在各个分辨率水平浏览基因组。主页的resource项下包含的简单重复序列标记搜索工具(SSR Candidate Marker Search Tool)可以用于对提交的序列进行SSR搜索,并利用PRIMER3搜索合适的扩增引物。用户可自行设定SSR类型和重复数目。SSRs广泛存在于各类生物基因组中,是应用较为广泛的遗传标记[21-22]。在基因组序列水平上对SSR的系统分析是SSR分子标记的基础性工作[23-24]。我 们 利 用 CPGR 数 据 库 的 SSR 数 据 对P.infestans与Pythium ultimum 基因组内的SSR进行了比较分析。P.infestans包含1958个SSR,平均每122.5kb有一个SSR,Pythium ultimum 包含1911个SSR,平均每22.39kb有一个SSR,两基因组内均以二、三、六碱基重复为最丰富,在基序的重复次数上也具有一定的相似性。在二、三、六碱基重复中,每种都存在优势基序,详见表2。在P.infestans基因组中,二、三、六碱基重复分别占总SSR的26.3%,21.81%和35.44%,在 Pythium ultimum中它们所占的百分比分别为27.68%,26.84%和27.42%,其他碱基重复所占的比例很少。在两基因组二、三碱基重复的优势基序中,大部分是相同的优势基序类型,但是六碱基重复的优势基序相同的比较少,而且,六碱基重复的基序类型分布也比较分散。但是两基因组的重复序列的长度都在20bp左右,长序列较少。而且有些基序类型没有出现。由以上分析可以看出,两个卵菌基因组内的SSR数目相差不多,但由于基因组大小差异导致SSR在基因组内的密度存在差异。优势碱基重复单元集中在二、三、六,说明其遗传变异速度中等。SSR长度比较集中。基因组水平上的SSR分析为在以后的研究中应用SSR标记提供了有用的信息。由于SSR标记在已测序卵菌基因组内的位置及侧翼序列是已知的,所以可以快速、准确地找到连锁的基因位点,从而完成候选基因的筛查、鉴定、基因注释及功能研究,SSR标记将在卵菌功能基因组研究中发挥重要作用。
CFGP(Comparative Fungal Genomics Platform)是一个综合性的真菌比较基因组学平台,包含283个真菌基因组的数据信息(6个卵菌)、专门数据库的超链接和各类比较基因组分析工具,包括BLAST、ClustalW、InterProScan、SignalP等[25]。平台采用数据驱动的用户界面(Data-driven User Interface DUI),用户可实现数据浏览,选择和分析的连续进行,节省收集数据、转化数据格式、提交数据、不同数据库间浏览查询的时间,极大地方便了大量数据的分析。该平台的数据查询主要是通过页面的SEQUENCE菜单实现,有10种查询方式:Taxonomy、Contig、Genome reference、MyGene、SWISSprot、MSIPI、NR、PDB、PDBchain、SequenceSet browser。数据通过 SNUGB(Seoul National University Genome Browser)以可视化方式显示,并可以选择显示不同的数据库信息。查询到的序列通过勾选进入到Favorites功能框架。Favorites作为生物信息分析的工作界面,包括Edit、Function、Anal-ysis和Download 4项功能,每个功能分若干项(见图2)。其中的Function包括BLAST、BLASTmatrix等27种分析工具,是主要的分析界面,可用于序列比对分析、功能结构域分析查询、系统进化分析、分泌蛋白分析、亚细胞定位、跨膜螺旋预测、RNA二级结构预测、翻译后修饰和保守结构域搜索。选择一种工具后,CFGP即通过PERL模块实现与相应程序的链接,设定参数即可运行获得结果。分析结果保存在History中。除了查看自己的分析结果,在History中也可查看其他人在CFGP分析的结果。在分析工具中,值得一提的是BLASTMatrix工具,它用于在多个不同真菌物种中搜索潜在的同源基因。BLASTMatrix结果提供提交序列在每个物种最佳命中序列的列表,该列表以每个物种的分类地位组织在一起,同时会提供在不同分类群组内同源基因的分布模式图。输出结果还包括InterPro或GO术语,可以帮助用户预测可能的基因功能,进一步分析确定其直系同源关系。
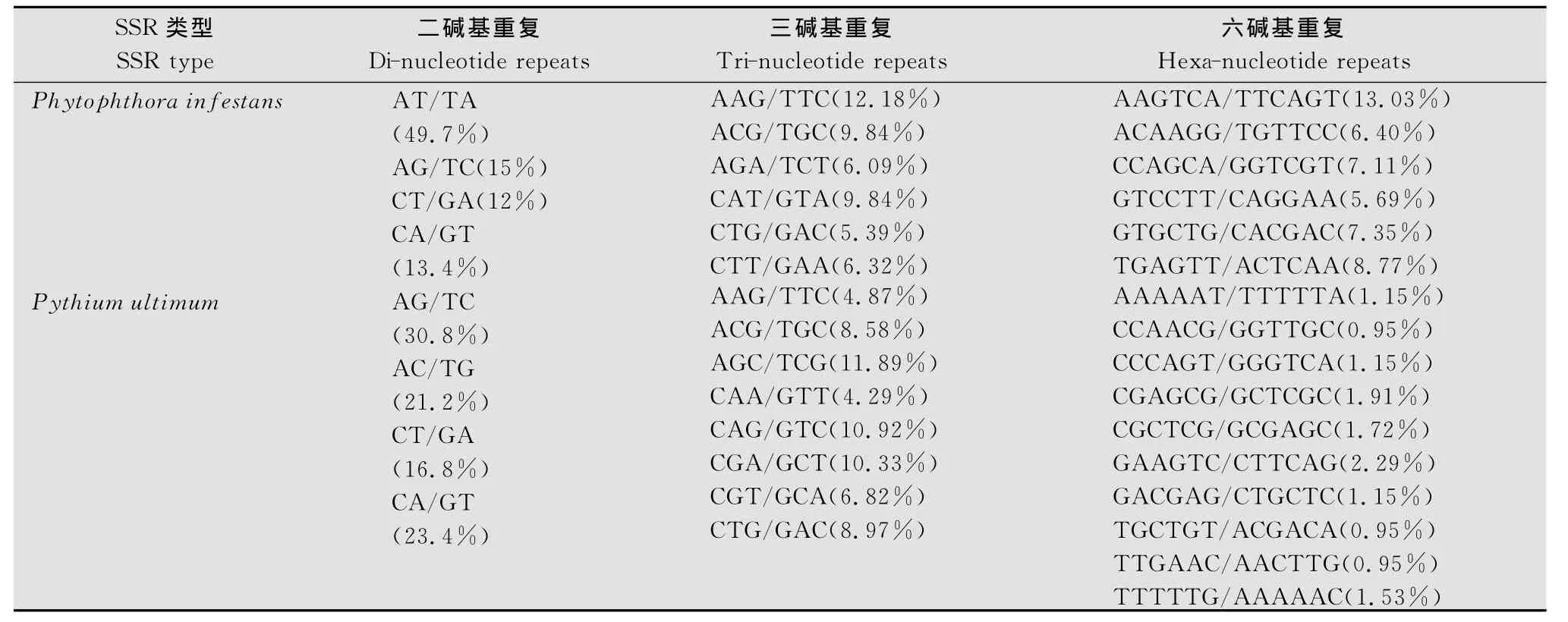
表2 P.infestans与Pythium ultimum基因组内SSR二、三、六碱基重复优势基序Tabel 2 Major motifs of di-,tri-and hexa-nucleotide repeats in genome of P.infestans and Pythium ultimum

图2 CFGP网站的Favorites工作界面Fig.2 The favorites interface in CFGP
FungiDB真菌功能基因组学资源,包括33种真菌和6种卵菌的基因组信息(V 2.1),整合了基因组序列和注释信息,可开展比较基因组学研究、基因表达分析、生物信息学分析和数据挖掘等[26]。FungiDB提供利用多种策略在选定的单个或多个物种中进行基因、ESTs、ORF、基因组序列等的快速检索,可实现多步检索策略的交叉、合并和删除,创建用户自己的检索策略进行数据分析。同时提供包括BLAST、序列检索(Sequence Retrieval)、文献检索(PubMed and Entrez)等分析工具。
2.3 卵菌专门数据库资源
除了综合性资源外,一些专门数据库也是卵菌功能基因组学研究的重要资源。专门数据库是针对特定目标由一级数据库衍生而来,对生物学知识和信息进一步整理形成的二级数据库,例如真菌分泌体数据库FSD,专门存储那些含有信号肽、在高尔基体或内质网进行加工能够分泌到细胞外的蛋白,这些蛋白通过在全基因组水平进行生物信息学分析确定,该数据库中可以查询到6种已测序卵菌的所有预测获得的分泌体蛋白[27]。另外,真菌的转录因子数据库FTFD[28]、真菌细胞色素 P450数据库 FCPD[29]等均存储有卵菌的相应蛋白的信息。这些专门数据库可用于对于特定基因或领域的研究。
3 总结
基因组学极大地提高了我们对于植物病原菌的理解,例如卵菌病原菌的大量测序揭示了调控宿主与寄生物相互作用的效应物分子类型[4,30-31]。通过在全基因组水平上对数据信息的挖掘,尤其是比较基因组学研究,将对揭示卵菌的特异性、病原菌群体结构、卵菌与病害关系、发现病原菌诊断标记起到重要作用[32]。随着测序方法的发展,数据产生规模的提高,对数据的处理和挖掘已经成为研究者的新挑战。
[1] Erwin D C,Riberro O K.Phytophthora diseases worldwide[M].St Paul:APS Press,1996:200-562.
[2] 郑小波.疫霉菌及其研究技术[M].北京:中国农业出版社,1997:1-29.
[3] Baldauf S L,Roger A J,Wenk-Siefert I,et al.A kingdom-level phylogeny of eukaryotes based on combined protein data[J].Science,2000,290(5493):972-977.
[4] Tyler B M,Tripathy S,Zhang X,et al.Phytophthora genome sequences uncover evolutionary origins and mechanisms of pathogenesis[J].Science,2006,313(5791):1261-1266.
[5] Haas B J,Kamoun S,Zody M C,et al.Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans[J].Nature,2009,461(7262):393-398.
[6] Lamour K H,Mudge J,Gobena D,et al.Genome sequencing and mapping reveal loss of heterozygosity as a mechanism for rapid adaptation in the vegetable pathogen Phytophthora capsici[J] Molecular Plant-Microbe Interactions,2012,25(10):1350-1360.
[7] Lévesque C A,Brouwer H,Cano L,et al.Genome sequence of the necrotrophic plant pathogen Pythium ultimumreveals original pathogenicity mechanisms and effector repertoire[J].Genome Biology,2010,11(7):R73-R94.
[8] Baxter L,Tripathy S,Ishaque N,et al.Signatures of adaptation to obligate biotrophy in the Hyaloperonospora arabidopsidis genome[J].Science,2010,330(6010):1549-1551.
[9] Tian M,Win J,Savory E,et al.454genome sequencing of Pseudoperonospora cubensis reveals effector proteins with a putative QXLR translocation motif[J].Molecular Plant-Microbe Interactions,2011,24(5):543-553.
[10]Lamour K H,Kamoun S.Oomycete genetics and genomics:Diversity,interactions and research tools[M].Hoboken,USA:Wiley-Blackwell Press,2009:540-582.
[11]Richards T A,Dacks J B,Jenkinson J M,et al.Evolution of filamentous plant pathogens:Gene exchange across eukaryotic kingdoms[J].Current Biology,2006,16(18):1857-1864.
[12]Kemen E,Gardiner A,Schultz-Larsen T,et al.Gene gain and loss during evolution of obligate parasitism in the white rust pathogen of Arabidopsis thaliana[J].PLoS Biology,2011,9(7):e1001094.
[13]Judelson H S.Dynamics and innovations within Oomycete genomes:Insights into biology,pathology,and evolution[J].Eukaryot Cell,2012,11(11):1304-1312.
[14]Grigoriev I V,Nordberg H,Shabalov I,et al.The genome portal of the department of Energy Joint Genome Institute[J].Nucleic Acids Research,2012,40(Database issue):D26-D32.
[15]Tripathy S,Pandey V N,Fang B,et al.VMD:A community annotation database for oomycetes and microbial genomes[J].Nucleic Acids Research,2006,34(Database issue):D379-D381.
[16]Links M G,Holub E,Jiang R H,et al.De novo sequence assembly of Albugo candida reveals a small genome relative to other biotrophic oomycetes[J].BMC Genomics,2011,12:503-514.
[17]Tripathy S,Deo T,Tyler B M.Oomycete transcriptomics database:A resource for oomycete transcriptomes[J].BMC Genomics,2012,13(1):303-316.
[18]Ye W,Wang X,Tao K,et al.Digital gene expression profiling of the Phytophthora sojae transcriptome[J].Molecular Plant-Microbe Interactions,2011,24(12):1530-1539.
[19]祁云霞,刘永斌,荣威恒.转录组研究新技术:RNA-Seq及其应用[J].遗传,2011,33(11):1191-1202.
[20]Hamilton J P,Neeno-Eckwall E C,Adhikari B N,et al.The comprehensive phytopathogen genomics resource:A webbased resource for data-mining plant pathogen genomes[J].Database:The Journal of Biological Databases and Curation,2011:bar53-65.
[21]Brurberg M B,Elameen A,Le V H,et al.Genetic analysis of Phytophthora infestans populations in the Nordic European countries reveals high genetic variability[J].Fungal Biology,2011,115(4-5):335-342.
[22]Vercauteren A,Larsen M,Goss E,et al.Identification of new polymorphic microsatellite markers in the NA1and NA2lineages of Phytophthora ramorum[J].Mycologia,2011,103(6):1245-1249.
[23]Schena L,Cardle L,Cooke D E.Use of genome sequence data in the design and testing of SSR markers for Phytophthoraspecies[J].BMC Genomics,2008,9:620-642.
[24]Garnica D P,Pinzón A M,Quesada-Ocampo L M,et al.Survey and analysis of microsatellites from transcript sequences in Phytophthoraspecies:Frequency,distribution,and potential as markers for the genus[J].BMC Genomics,2006,7:245-255.
[25]Park J,Park B,Jung K,et al.CFGP:A web-based,comparative fungal genomics platform[J].Nucleic Acids Research,2008,36(Database issue):D562-D571.
[26]Stajich J E,Harris T,Brunk B P,et al.FungiDB:An integrated functional genomics database for fungi[J].Nucleic Acids Research,2012,40(Database issue):D675-D681.
[27]Choi J,Park J,Kim D,et al.Fungal Secretome Database:Integrated platform for annotation of fungal secretomes[J].BMC Genomics,2010,11:105-119.
[28]Park J,Park J,Jang S,et al.FTFD:An informatics pipeline supporting phylogenomic analysis of fungal transcription factors[J].Bioinformatics,2008,24(7):1024-1025.
[29]Park J,Lee S,Choi J,et al.Fungal cytochrome P450database[J].BMC Genomics,2008,9:402-412.
[30]Win J,Kamoun S.Adaptive evolution has targeted the C-terminal domain of the RXLR effectors of plant pathogenic oomycetes[J].Plant Signal Behavior,2008,3(4):251-253.
[31]Savory E A,Zou C,Adhikari B N,et al.Alternative splicing of a multi-drug transporter from Pseudoperonospora cubensis generates an RXLR effector protein that elicits a rapid cell death[J].PLoS One,2012,7(4):e34701-e34713.
[32]Seidl M F,Van den Ackerveken G,Govers F,et al.A domain-centric analysis of oomycete plant pathogen genomes reveals unique protein organization[J].Plant Physiology,2011,155(2):628-644.
