Zr/Ti摩尔比对锶锆钛复合氧化物在可见光下光催化性能的影响
2013-06-23黄海凤贾建明卢晗锋张宏华潘烈群
黄海凤 贾建明 卢晗锋 张宏华 潘烈群
(1浙江工业大学生物与环境工程学院,杭州310014;2浙江工业大学化学工程与材料科学学院,催化反应工程研究所,杭州310014;3富阳市环境保护监测站,浙江富阳311400)
1 引言
半导电性材料多相光催化技术是近年来的研究热点,1其中,ABO3钙钛矿型复合氧化物因其在磁性、电导性、催化降解染料废水等方面的优异性能受到了国内外学者的广泛关注.2-4SrTiO3钙钛矿是一种最具应用前景的光催化材料,在分解水制氢、5,6光降解有机污染物7,8等方面均有良好表现.但SrTiO3禁带宽度为3.2 eV左右,9在紫外光下才能激发光生电子,其应用受到了很大的限制.于是对SrTiO3进行改性,拓宽其光响应范围,提高量子效率成为光催化研究工作者追求的目标.目前主流的改性方法集中于离子掺杂10-13与贵金属沉积,14,15而多种氧化物复合改性ABO3钙钛矿却鲜有报道.另一方面,TiO2研究领域中通过多种氧化物复合改性均取得了较大的成功.16-18
本课题组19前期考察了SrTiO3的制备方法,发现分步沉积法得到的SrTiO3催化剂活性最高.为了进一步探索有效的可见光催化剂,论文设计以分步沉积法制备不同Zr/Ti摩尔比的锶锆钛(SZT)复合氧化物催化剂.用XRD、SEM、TEM、UV-Vis对这些催化剂的结构形态进行了表征,讨论了不同Zr/Ti摩尔比对其可见光下的光催化性能影响.
2 实验部分
2.1 试剂
钛酸四丁酯(分析纯98%),上海美兴化工股份有限公司;硝酸锶(分析纯99.5%),上海恒信化学试剂有限公司;五水硝酸锆(分析纯99.8%),国药集团化学试剂有限公司;氨水(分析纯25%),杭州长征化学试剂有限公司;亚硝酸钠(分析纯99%),上海振欣试剂厂;指示剂亚甲基蓝(指示剂82%),浙江温州市东升化工试剂厂;无水乙醇(分析纯99.7%);上海海逸科贸有限公司.
2.2 催化剂制备
取一定量钛酸四丁酯分散于乙醇中,并滴加氨水使pH值约为8,得到乳白色混合液,将混合液抽滤后得到凝胶状白色沉淀;称取一定量的五水硝酸锆溶于水中,与适量的硝酸锶配成的溶液混合,再与抽滤后的凝胶状白色沉淀混合,磁力搅拌,旋转蒸干,后放入110°C干燥箱干燥17 h,研磨后放置马弗炉中分段焙烧,400°C焙烧4 h,700°C焙烧4 h,高温焙烧下得到完善的晶型.在定量加入硝酸锶条件下,根据钛酸四丁酯和硝酸锆的不同摩尔比,分别得到样品SZT-X,记为SZT-2/8、SZT-3/7、SZT-4/6、SZT-5/5、SZT-8/2.
2.3 催化剂表征
XRD表征采用ARL SCINTAG XTRA X射线衍射仪分析样品的物相,Ni滤波,使用Cu靶Kα辐射,工作电压为45 kV,工作电流为40 mA,扫描范围2θ=10°-80°.BET(Brunauer-Emmett-Teller)表征采用Micromeritics ASAP 2010C型吸附仪测定,样品测定前经250°C脱气处理4 h.SEM表征采用PHILIPS公司生产的Philips XL-30扫描电子显微镜及能谱仪测定,扫描前进行喷金处理.TEM表征采用荷兰FEI公司生产TecnaiG2 F30高分辨透射电子显微镜及能谱分析仪测定,测定前进行超声分散样品处理.UV-Vis表征采用日本SHIMADZU公司生产的Spec TM-BWS003光谱仪测试,以BaSO4为参比.
2.4 光催化降解实验
光催化降解反应在自制的圆柱形耐热石英反应器中进行,为排除外界光源的干扰,光催化反应在暗箱中进行.反应器中心为镝灯(400 W),置于2 mol·L-1的亚硝酸钠溶液中.亚硝酸钠溶液将光源散发出的400 nm以下的光阻隔,得到可见光.夹层通循环水控制反应温度,反应保持在25°C,采用亚甲基蓝作为降解对象.将500 mL浓度为10 mol·L-1的亚甲基蓝水溶液注入自制的套层玻璃反应器中,催化剂用量为2.0 g·L-1.在磁力搅拌下,先暗吸附20 min,后打开镝灯(400 W),光稳定10 min后,开始记录光照反应时间,每隔一定的时间取样,用紫外-可见分光光度计(岛津UV-2660)在650 nm处测其吸光度的变化,由此计算亚甲基蓝的脱色率.
暗箱吸附实验在相同反应器中进行,无光条件,反应40 min,每隔5 min取样.用紫外-可见分光光度计在650 nm处测其吸光度的变化,由此计算亚甲基蓝的脱色率.
3 结果与讨论
3.1 SZT可见光光催化活性分析
图1为不同Zr/Ti摩尔比的锶锆钛(SZT)复合氧化物催化剂的暗箱吸附实验结果.我们定义A0和A分别为0和t时染料溶液的吸光度,以D=(1-A/A0)×100%表示t时刻锶锆钛催化剂对染料溶液亚甲基蓝的降解效果.结果显示,催化剂吸附对于亚甲基蓝的降解最终效果均在5%以下.且样品暗箱吸附20 min之后,催化剂对于亚甲基蓝基本不再吸附降解.

图1 不同Zr/Ti摩尔比的锶锆钛催化剂(SZT-X)对亚甲基蓝的暗箱吸附作用Fig.1 Adsorption of methylene blue in black box by Sr-Zr-Ti mixed oxide catalysts(SZT-X)with different Zr/Ti molar ratios
实验结果排除了“暗箱吸附20 min,光照稳定10 min后开始计数的光催化反应”中吸附作用的影响.我们定义可见光下SZT-X催化剂催化降解亚甲基蓝的能力表示催化剂活性大小.图2与图3为可见光照射下SZT-X催化剂催化降解亚甲基蓝的活性数据比较.速率常数(k)计算采用一级动力学方程式:20,21
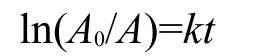
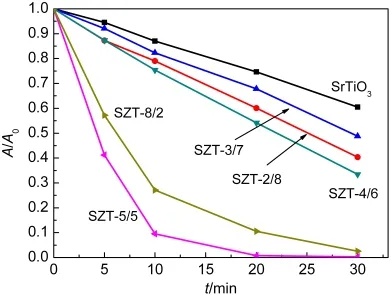
图2 可见光下不同Zr/Ti摩尔比的锶锆钛催化剂(SZT-X)对亚甲基蓝的降解作用Fig.2 Decolorization ration of methylene blue on visible light by Sr-Zr-Ti mixed oxide catalysts(SZT-X)with different Zr/Ti molar ratios
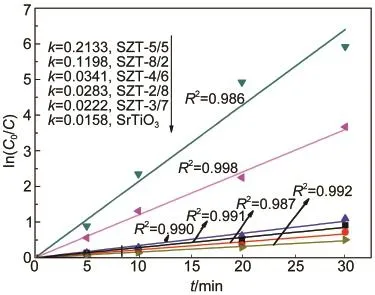
图3 可见光下不同Zr/Ti摩尔比的锶锆钛催化剂(SZT-X)降解亚甲基蓝的一级反应速率Fig.3 First-order reaction rates of Sr-Zr-Ti mixed oxide catalysts(SZT-X)with different Zr/Ti molar ratios for the photodegradation of methylene blue on visible light
结果显示,当Zr/Ti摩尔比<1时,样品的一级反应速率常数在同一个数量级上,活性优于纯SrTiO3但提高幅度不大;当Zr/Ti摩尔比≥1时,样品的一级反应速率常数与纯SrTiO3样品相比,提高了几个数量级,其中,SZT-5/5催化剂表现出最高的光催化活性,其一级反应速率常数是SZT-8/2的1.8倍,达到0.2133 min-1,是同等光照条件下纯SrTiO3样品(0.0158 min-1)的13.5倍.说明不同Zr/Ti摩尔比的锶锆钛(SZT)复合氧化物催化剂能不同程度地提高SrTiO3样品的光催化活性,从光催化降解实验结果看,Zr的最佳掺入量为n(Zr):n(Ti)=1:1.光催化活性的比较结 果 为 SZT-5/5>SZT-8/2>>SZT-4/6>SZT-2/8>SZT-3/7>纯SrTiO3.
3.2 XRD表征分析
图4(a)为SZT-X催化剂的XRD表征.由图可知,Zr/Ti摩尔比<1时,SrTiO3结晶度急剧下降,Sr2TiO4与SrTiO3晶相比逐步增大.而当Zr/Ti摩尔比≥1时,SZT-X催化剂出现了新的晶相SrZrO3与TiO2.
图4(b)为Zr/Ti摩尔比<1时SZT-X催化剂的特征峰XRD图谱(2θ范围为30.8°-33.5°).发现在Zr/Ti摩尔比<1条件下,随着Zr含量的增多,SZT-X催化剂的SrTiO3的晶相逐步被Sr2TiO4取代,左侧Sr2TiO4特征峰逐渐向右偏移.根据现代晶体学理论,22,23当杂质和基质的离子半径和电负性相接近时,形成杂质替位的几率较大,晶相图谱表现为同一方向微弱的移动,晶格间距膨胀或缩小.已知Zr的离子半径与元素的相对电负性都和Ti比较接近:Ti的有效离子半径为0.0605,24配位数为6,电负性(鲍林标度)为1.54;Zr的有效离子半径为0.072,配位数为6,电负性(鲍林标度)为1.33.
表1为Zr/Ti摩尔比<1条件下的SZT-X催化剂的物理性质参数,由表可知,在Zr/Ti摩尔比<1条件下,样品的晶格参数随Zr/Ti摩尔比的增大而有微小变大,晶格间距也稍变大.这表明Zr/Ti摩尔比<1条件下,SZT-X催化剂的Ti4+与Zr4+在高温条件下发生了同质取代,由于Zr4+的有效离子半径大于Ti4+,会影响周围O2-的排布情况,产生晶格缺陷.而半导体晶格缺陷的产生将降低SZT-X催化剂的化学稳定性,提高其光催化活性.

图4 不同Zr/Ti摩尔比的锶锆钛催化剂(SZT-X)的XRD谱图Fig.4 XRD patterns of Sr-Zr-Ti mixed oxide catalysts(SZT-X)with different Zr/Ti molar ratios
图4(c)为Zr/Ti摩尔比≥1时SZT-X催化剂的特征峰 XRD 图谱(2θ范围为 22.5°-33°).由图可知,SZT-5/5中Sr2TiO4与SrTiO3晶相完全消失,出现了以 SrZrO3与 TiO2、亚氧化钛 TinO2n-1(n=4,9)为特征峰的晶相结构.而SZT-8/2催化剂在2θ=30.297°处形成了ZrO2晶相,特征峰SrZrO3相比SZT-5/5样品向右移动了0.025°.值得注意的是,在2θ=25.21°(TiO2特征峰)处附近,出现了大量的亚氧化钛TinO2n-1(n=4,9)晶相(Ti4O7与Ti9O17).亚氧化钛TinO2n-1(n=4,9)晶相是一系列缺氧钛氧化物的统称,一方面具有TiO2的一些优良特性,比如优异的抗氧化性、25良好的抗电化学腐蚀性26等特点,另一方面还具有无磁性、高电导率、27,28光学性能独特29等优点.特别是Magnéli相亚氧化钛Ti4O7具有类金属的高导电性,禁带宽度仅为零点几电子伏特.30,31
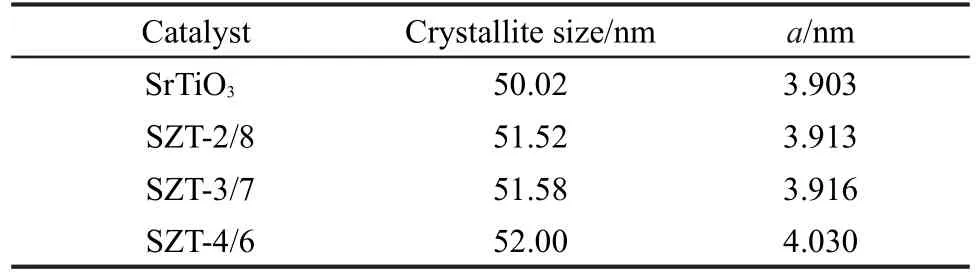
表1 Zr/Ti摩尔比小于1时锶锆钛催化剂(SZT-X)的晶粒尺寸与晶胞参数(a)Table 1 Crystal size and lattice parameter(a)of Sr-Zr-Ti mixed oxide catalysts(SZT-X)with Zr/Ti molar ratio<1
图5为各种半导体化合物能带结构对应关系,32由图可知在可见光区域,SrZrO3的价带电子能发生跃迁产生光生电子和空穴.结合XRD图谱分析,亚氧化钛TinO2n-1(n=4,9)晶相的存在促使催化剂样品SZT-5/5与SZT-8/2形成了低于Ti4+的空穴捕获中心.光催化过程中,分步沉积法制备的具有“核壳”结构的样品受光照射产生的电子很快转移到亚氧化钛TinO2n-1(n=4,9),有效降低了电子与空穴的复合几率.在Zr/Ti摩尔比≥1条件下,光催化活性得到了几个数量级的提高.图6为SZT-X(X≥1)催化剂光降解亚甲基蓝的反应模型示意图.
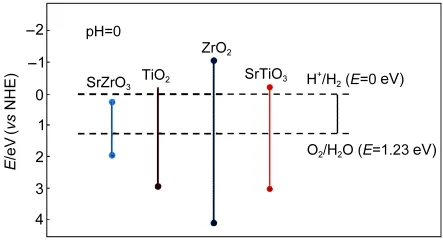
图5 在pH=0时各种半导体化合物能带结构对应关系Fig.5 Band structure of different semiconductor compound with pH=0
在图4(c)中,SZT-8/2特征峰相比SZT-5/5出现了ZrO2晶相,根据图5可知,ZrO2的禁带宽度为5.5 eV,较高的带隙能阻碍了光生电子和空穴的传输,提高了其复合几率;另一方面,随着Zr掺入量得进一步增大,亚氧化钛TinO2n-1(n=4,9)晶相(Ti4O7与Ti9O17)的结晶度都有一定程度的下降.所以在Zr/Ti摩尔比≥1的条件下,能较大提高SrTiO3样品的光催化活性,但Zr含量过量时,样品的光催化活性下降.
3.3 透射电镜表征分析
图7为SZT-X催化剂的TEM表征结果.结果表明,不同Zr/Ti摩尔比的SZT-X催化剂均显示出良好的晶体结构,晶格间距与XRD计算数值相吻合.当Zr/Ti摩尔比<1时,晶格间距随着Zr掺入量的增多而逐渐增大.其中,SZT-2/8催化剂样品存在点缺陷;SZT-3/7和SZT-4/6催化剂样品存在线缺陷.23该结果与图4(b)XRD分析相吻合,进一步解释了少量Zr掺入的SZT-X光催化剂活性提高的原因.随Zr/Ti摩尔比的进一步增大,晶格间距发生了突变.通过SZT-5/5的EDX表征,发现其样品表面的Sr与Zr的含量明显高于Ti含量.对比文献报道,我们认为此时SZT-5/5催化剂出现了新的晶相SrZrO3与TiO2及一系列缺氧钛氧化物.随着Zr掺入量的不断增加,当X=8/2,可以看到SrZrO3逐渐转变为ZrO2.这一结果与图4(c)XRD分析结果一致.
3.4 扫描电镜与BET表征分析
图8为SZT-X催化剂的SEM表征结果.结果显示,不同Zr/Ti摩尔比的样品颗粒大小和表面形貌具有明显差异.比较SrTiO3催化剂,当Zr/Ti摩尔比<1时,样品的颗粒较为明显,呈现两种颗粒形貌,即细小颗粒(约30 nm)和较大颗粒(约90 nm),这可能与样品同时具有SrTiO3与Sr2TiO4两种晶相的结构特征有关.随着Zr含量的增多,样品颗粒团聚现象明显,样品颗粒尺寸增大,符合样品BET表征结果与XRD谢乐公式计算结果,见表2.随着Zr/Ti摩尔比的进一步增大,样品形貌发生了明显的变化,颗粒呈现块状.其中SZT-5/5的样品整体性最好,样品表面像被包裹了一层致密的薄膜,对比文献发现,样品表面可能是SrZrO3.33Zr含量进一步增大,可以看出表面薄膜已消失,暴露出一定的立方结构物质.
一般来说,光催化剂具有较小的晶粒及较大的比表面积,对光催化反应有利.而从BET表征结果看出SZT-2/8具有最大的比表面积,我们推测较大的比表面积具有更多的活性点位,并最终导致光催化活性SZT-2/8>SZT-3/7.
3.5 紫外漫反射光谱UV-VIS分析
图9为SZT-X催化剂的紫外-可见漫反射光谱.结果显示,当Zr/Ti摩尔比<1时,样品的光吸收带发生了明显的红移,随着Zr/Ti摩尔比的增大,其对可见光的吸收强度不断增强,其中SZT-4/6在可见光区域具有最高的吸收强度.结合XRD与TEM表征结果,Zr的同质取代有利于提高样品对可见光的吸收强度.所以在Zr/Ti摩尔比<1条件下,样品光催化活性应随着Zr/Ti摩尔比的增大而增强.但由于样品比表面积SZT-2/8>SZT-3/7,造成了SZT-2/8催化剂单位面积活性点位的数目大于催化剂SZT-3/7,所以导致光催化活性催化剂SZT-2/8>SZT-3/7;而虽然比表面积催化剂SZT-4/6>SZT-3/7,但由于SZT-4/6的可见光吸收强度远大于SZT-3/7,同样造成单位面积活性点位数目催化剂SZT-4/6>SZT-3/7,所以光催化活性催化剂SZT-4/6>SZT-2/8>SZT-3/7.

图6 锶锆钛催化剂(SZT-X)(X≥1)催化剂光降解亚甲基蓝反应模型示意图Fig.6 Possible model of photocatalytic degradation of methylene blue of Sr-Zr-Ti mixed oxide catalysts SZT-X(X≥1)

图7 不同Zr/Ti摩尔比的锶锆钛催化剂(SZT-X)的TEM图像Fig.7 TEM images of Sr-Zr-Ti mixed oxide catalysts(SZT-X)with different Zr/Ti molar ratios

图8 不同Zr/Ti摩尔比的锶锆钛催化剂(SZT-X)的SEM图像Fig.8 SEM images of Sr-Zr-Ti mixed oxide catalysts(SZT-X)with different Zr/Ti molar ratios
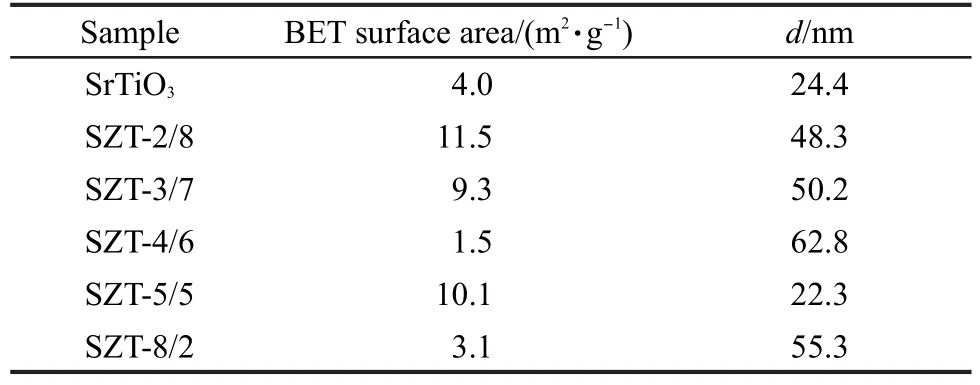
表2 样品晶粒大小(d)及比表面积Table 2 Crystallite size(d)and surface area of samples
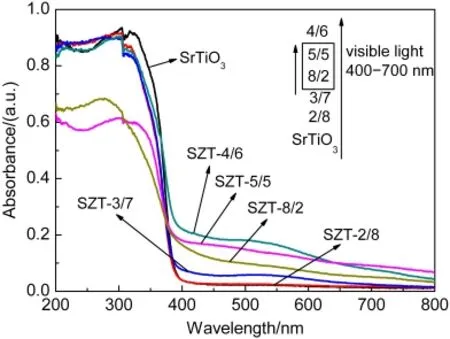
图9 不同Zr/Ti摩尔比的锶锆钛催化剂(SZT-X)的紫外可见漫反射光谱Fig.9 UV-Vis diffuse reflection spectra of Sr-Zr-Ti mixed oxide catalysts(SZT-X)with different Zr/Ti molar ratios
当Zr/Ti摩尔比≥1时,SZT产生SrZrO3/TinO2n-1(n=4,9)的新晶相,不同的晶相结构对可见光的吸收强度不同,但图9结果显示其吸收强度同样增强,且在紫外光区域的吸收值发生了突降,结合图5分析,可知在新的混合催化晶相中,SrZrO3的减少不利于样品对可见光的吸收.所以Zr/Ti摩尔比≥1时,光催化活性SZT-5/5优于SZT-8/2.
因此,催化剂活性不仅取决于催化剂的比表面积以及光响应范围,更取决于催化剂的晶相结构,能否有效促进电子与空穴的分离.
4 结论
通过分步沉积法制备了不同Zr/Ti摩尔比的锶锆钛(SZT)复合氧化物催化剂.对催化剂的表征及催化活性研究显示,Zr/Ti摩尔比<1时催化剂出现类质同象,催化剂产生晶格缺陷且光催化活性得到小幅提高;当Zr/Ti摩尔比≥1时,催化剂会产生以SrZrO3、TiO2和TinO2n-1(n=4,9)为主的新晶相.TinO2n-1(n=4,9)晶相易形成低于Ti4+的空穴捕获中心,有效降低了电子与空穴的复合几率,并大幅提高催化剂的可见光光催化活性.其中,SZT-5/5表现出最高的光催化活性,其一级反应速率常数达到0.2133 min-1,是同等光照条件下SrTiO3样品(0.0158 min-1)的13.5倍.
(1)Yan,X.R.;Li,X.H.;Huo,M.L.;Guo,W.W.;Gong,Y.J.Acta Phys.-Chim.Sin.2001,17,23.[颜秀如,李晓红,霍明亮,郭伟巍,巩永进.物理化学学报,2001,17,23.]doi:10.3866/PKU.WHXB20010105
(2)Kurokawa,H.;Yang,L.M.;Jacobson,C.P.;Jacobson,C.P.;De Jonghe,L.C.;Visco,S.J.Power Sources2007,164(2),510.doi:10.1016/j.jpowsour.2006.11.048
(3) Huang,H.F.;Tang,W.;Chen,Y.F.;Chen,B.F.J.Mol.Catal.2005,19(5),351.[黄海凤,唐 伟,陈银飞,陈碧芬.分子催化,2005,19(5),351.]
(4) Lee,M.S.;Meyer,J.U.Sensors and Actuators B,Chemical2000,68(1-3),293.doi:10.1016/S0925-4005(00)00447-0
(5) Luo,W.J.;Li,Z.S.;Jiang,X.J.;Yu,T.;Liu,L.F.;Chen,X.Y.;Ye,J.H.;Zou,Z.G.Phys.Chem.Chem.Phys.2008,10,6717.doi:10.1039/b803996h
(6) Puangpetch,T.;Sreethawong,T.;Chavadej,S.Int.J.Hydrog.Energy2010,35,6532.
(7)Wang,D.;Kako,T.;Ye,J.J.Phys.Chem.C2009,113,3785.doi:10.1021/jp807393a
(8) Chen,L.;Zhang,S.C.;Wang,L.Q.;Xue,D.F.;Yin,S.J.Crystal Growth2009,311,746.
(9) Ryoko,K.;Tatsuya,I.;Hideki,K.;Akihiko,K.J.Phys.Chem.B2004,108,8992.doi:10.1021/jp049556p
(10) Onishi,T.J.Top Catal.2010,53,566.doi:10.1007/s11244-010-9488-6
(11) Galinetto,P.;Casiraghi,A.;Mozzati,M.C.;Azzoni,C.B.;Norton,D.;Boatner,L.A.Ferroelectrics2008,368(1),120.doi:10.1080/00150190802368248
(12)Hua,N.P.;Wu,Z.Y.;Du,Y.K.;Zou,Z.G.;Yang,P.Acta Phys.-Chim.Sin.2005,21(10),1085.[华南平,吴遵义,杜玉扣,邹志刚,杨 平.物理化学学报,2005,21(10),1085.]doi:10.3866/PKU.WHXB20051004
(13)Lu,H.F.;Zhou,Y.;Xu,B.Q.;Chen,Y.F.;Liu,H.Z.Acta Phys.-Chim.Sin.2008,24(3),459.[卢晗锋,周 瑛,徐柏庆,陈银飞,刘化章.物理化学学报,2008,24(3),459.]doi:10.3866/PKU.WHXB20080319
(14) Puangpetch,T.;Sreethawong,T.;Chavadej,S.Int.J.Hydrog.Energy2010,35,6531.doi:10.1016/j.ijhydene.2010.04.015
(15)Wei,W.;Dai,Y.;Guo,M.;Zhu,Y.Z.;Huang,B.B.J.Phys.Chem.C2010,114,10917.
(16) Yang,J.;Li,D.;Wang,X.J.Solid State Chem.2002,165(1),193.doi:10.1006/jssc.2001.9526
(17) Tanaka,T.;Teramura Kentaro,Y.T.J.Photochem.Photobiol.A-Chem.2002,148(1-3),277.doi:10.1016/S1010-6030(02)00054-0
(18) Kataoka,S.;Tompkins,D.T.;Zeltner,W.A.J.Photochem.Photobiol.A-Chem.2002,148(1-3),323.doi:10.1016/S1010-6030(02)00059-X
(19) Pan,L.Q.;Lu,H.F.;Huang,H.F.Rare Earths2011,3(29),284.[潘烈群,卢晗锋,黄海凤.中国稀土学报,2011,3(29),284.]
(20)Yu,C.L.;Fan,C.F.;Yu,J.M.Mater.Res.Bull.2011,46(1),145.
(21) Tennakone,K.;Ileperuma,O.A.;Bandara,J.M.S.;Kiridena,W.C.B.Semicond.Sci.Technol.1992,7(3),424.doi:10.1088/0268-1242/7/3B/109
(22)Wei,G.P.;Jiang,C.H.;Zhen,W.;Jin,D.R.Crystal Structure and Defects,1st ed.;Code of Practice for Design and Construction:Beijing,2010;pp 5,64-120.[魏光普,姜传海,甄 伟,金灯仁.晶体结构与缺陷,第一版.北京:中国水利水电出版社;2010:5,64-120.]
(23) Chen,J.Z.Modern Crystal Chemistry,1st ed.;Science and Technology Press:Beijin g,2010;pp 140-160.[陈敬中.现代晶体化学,第一版.北京:科学技术出版社,2010:140-60.]
(24) Xu,Y.L.Basic of Oxide and Compound Semiconductor,1st ed.;Xidian University Press:Xiʹan,1991.[徐毓龙.氧化物与化合物半导体基础,第一版.西安:西安电子科技大学出版社,1991.]
(25) Han,W.Q.;Wang,X.L.Appl.Phys.Lett.2010,2(12),3709.
(26) Walsh,F.C.;Wills,R.G.A.Electrochimica Acta2010,55,6342.doi:10.1016/j.electacta.2010.05.011
(27)Chen,G.Y.;Simon,R.B.;Thomas,E.M.The Electrochemical Society2002,149(8),A1092.
(28) Lakkis,S.;Schlenkcr,C.;Chakravcrty,B.K.;Buder,R.;Marezio,M.Physical Review B1976,14(4),1429.
(29) Masayuki,W.;Wakana,U.;Tetsusuke,H.Luminescence2007,122-123,393.
(30) Leandro,L.;Giuseppe,M.Physical Review B2009,79,245133.doi:10.1103/PhysRevB.79.245133
(31) Eyert,V.;Schwingenschl,U.;Eckern,U.Chemical Physics Letters2004,390,151.doi:10.1016/j.cplett.2004.04.015
(32) Xu,Y.;Schoonen,M.A.A.American Mineralogist2000,85,543.
(33)Liu,S.W.;Lu,M.K.;Song,C.F.;Wang,S.F.;Gu,F.;Cheng,X.F.;Xu,D.;Yuan,D.R.J.Functional Materials2004,35(2),233.[刘素文,吕孟凯,宋春风,王淑芬,顾 锋,程秀风,许 东,袁多荣.功能材料,2004,35(2),233.]
