玻恩-哈伯循环在无机化学中的应用
2013-02-13何丽君李生英徐飞许世红霍彩霞
何丽君 李生英 徐飞 许世红 霍彩霞
(兰州城市学院化学与环境科学学院 甘肃兰州730070)
一个化学反应从始态到终态的实际历程可能比较复杂,甚至不能直接进行,但可以设计一个分步的、甚至是虚构的途径。尽管设计的途径和实际途径往往不同,但由于过程的焓变ΔH是状态函数,其总的热效应是相同的,与反应途径无关;基于上述思想,德国物理学家Max Born(麦克斯·玻恩)和德国化学家Fritz Haber(弗里茨·哈伯)设计了一种循环,可以用来进行各种热化学数据的简单计算,起到验证和补充实验数据的作用,即玻恩-哈伯循环。玻恩-哈伯循环在无机化学中有着广泛应用,除用于计算晶格能外,还可以求算元素的电子亲和能和质子亲和能,计算离子化合物的生成焓,从而指导无机物的合成,特别是用于探讨无机化合物的性质规律。玻恩-哈伯循环已成为学习和研究无机化学所必需的基础理论知识和有力的工具。本文就其在无机化学中的一些应用作简单介绍。
1 晶格能与玻恩-哈伯循环
1.1 求算晶格能[1]
晶格能(U)是指将1mol离子晶体里的正、负离子(克服晶体中的静电引力)完全气化而远离所需要的能量。其数值可由实验方法得到,但由于实验技术上的困难,目前大多数离子晶体的晶格能是利用1919年玻恩和哈伯设计的玻恩-哈伯循环间接测定。如求算NaCl晶体的晶格能,按照定义,即过程(1)所需要的能量。

玻恩-哈伯循环把过程(1)分解为下面若干过程:

其中过程a的能量变化E(a)等于由单质化合生成离子晶体的生成热()的负值;过程b的能量变化E(b)为1mol金属钠气化吸收的能量(升华热,S)与0.5mol氯分子的解离能(D)之和;过程c的能量变化E(c)为金属钠的第一电离能(Ⅰ)与氯原子的第一电子亲和能(Y)之和。晶格能就是上述能量项的加和:

由于以上各能量项均可用实验方法测定,所以这种由玻恩和哈伯设计的热化学循环可估算出许多离子晶体的晶格能。若经计算得出某种离子晶体的晶格能为负值,则意味着这种离子晶体不可能生成。
通过这种晶格能的求算,还可以帮助我们深入了解玻恩-哈伯循环中各个热力学参数在化学键合中的相对重要性。如在上述热化学循环中,E(a)恒为正值,它与其他能量项相比数值较小,而且对于不同的物质其值相差不大。钠的第一电离能为正值,Cl的第一电子亲和能为负值,二者总和是正值,总结果U>0,说明离子间的相互作用产生的晶格能使得离子晶体稳定存在。
1.2 求算电子亲和能
将晶格能计算与玻恩-哈伯循环结合起来可以计算循环中的任一热力学参数。例如,多年来电子亲和能的测定比较困难,但可用玻恩-哈伯循环求得。表1为通过KCl、RbCl及CsCl的晶格能求算氯原子的电子亲和能的实例。

表1 氯原子的电子亲和能
通过以上计算可以看出,分别由KCl、RbCl和CsCl的晶格能通过玻恩-哈伯循环所得氯原子的电子亲和能是一致的。
再如利用MgO的晶格能可求氧原子形成O2-的电子亲和能:

计算得到Y(O)=693kJ·mol-1,可见这一过程为吸热过程。氧原子电子亲和能为正值,这是因为要迫使第2个电子与带负电荷的O-结合需要供给能量的缘故。
可见,利用玻恩-哈伯循环可求算元素原子的电子亲和能。
1.3 质子亲和能的求算[2]
在分子离子反应中,分子争夺质子时(质子化时)释放出的能量称为质子亲和能,常用P表示。如想知道气态NH3(过程(2))结合H+形成的亲和能,可设计成如下的玻恩-哈伯循环,利用NH4X(卤化铵)的晶格能求算。

玻恩-哈伯循环把过程(2)分解为下面若干过程:

根据盖斯定律:

可计算出NH3的质子亲和能P。对应于NH4Cl、NH4Br、NH4I,P(NH3)分别为-894.55、-894.95、-886.18kJ·mol-1。用同样的方法可以计算出许多分子或离子的质子亲和能。

表2 部分物质的质子亲和能
NH3(g)对H+(g)的亲和能也可看作是(g)络离子中NH3对H+亲和能。同样可用类似的方法计算某些络离子如等的BF3(g)对F-(g)的亲和能。
1.4 指导无机化合物的合成
应用晶格能和玻恩-哈伯循环可以计算出一些未知离子化合物的生成焓或反应焓,用来预计合成某些无机化合物的可能性。如英国化学家巴特列利用这个理论方法作指导,于1962年8月成功地合成了第一个惰性气体化合物XePtF6。
下面通过近似计算进一步定量地说明晶格能和玻恩-哈伯循环对指导合成XePtF6的重要意义。
首先通过卡普斯钦斯基公式[2]计算XePtF6的晶格能为458.7kJ·mol-1,比O2PtF6的晶格能(506kJ·mol-1)稍小一些。
再根据玻恩-哈伯循环计算由Xe与PtF6反应生成XePtF6的热效应。

已知:U(XePtF6)=458.7kJ·mol-1,Y(PtF6)=-771kJ·mol-1,Ⅰ(Xe)=1170kJ·mol-1,则=-59.7kJ·mol-1。XePtF6的生成热为负值,说明生成XePtF6的反应是放热反应,预计有可能生成XePtF6,从相关数据中还可以看出PtF6的电子亲和能大是XePtF6得以合成的主要原因。
2 计算离子键的键能
离子键的键能是指在标准状态下,将1mol的气态AB分子中的化学键断开,离解为中性气态原子A和B所需的能量。如NaCl键能的求算,按照定义,应该是过程(3)的能量变化E。


其中E(a)为气态离子生成气态NaCl分子时放出能量的负值;E(b)为金属钠的第一电离能(Ⅰ)的负值与氯原子的第一电子亲和能(Y)的负值之和。
已知:E(a)=526kJ·mol-1;E(b)=-128kJ·mol-1;则E=398kJ·mol-1,故气态NaCl的键能为398kJ·mol-1。
由于离子型物质一般以晶体状态存在,所以离子键的键能数据并不常用,而通常用晶格能大小来衡量离子键的强弱。但通过讨论可知NaCl(s)的晶格能与其键能是两个不同的概念。
3 金属活泼性与玻恩-哈伯循环
金属活动顺序表是通过金属电对的φӨ得到的。金属的标准电极电势越小,就越活泼,在水溶液中还原性也越强。φӨ的大小又由哪些因素决定呢?也可通过玻恩-哈伯循环来分析。对于金属失电子这一过程可写成如下通式:

秦观五十篇策论历来评价甚多,对其策论成就的看法却是呈两极化的。褒之者认为其“灼可资庙谋”[1]19,具有实际功用,秦观也被目为“国士无双”[1]20,堪为大用。贬之者则认为,秦观策论仅限为党争而作,未有实际功用,行文也是结构单一的应试之作。关于秦观策论实用价值与文学价值的探讨,前人论述已备,本是无缘置喙。然本文仍不揣浅陋,希望从另一种视角来探讨秦观策论文的价值,以期有所发现。
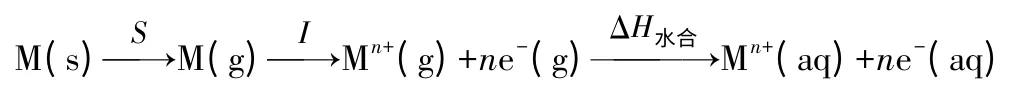

表3 铜族原子转为M+(aq)时的能量变化
从表3可以看出,按铜、银、金顺序,M(s)→M+(aq)过程所需总能量越来越大,即单质形成M+(aq)的活泼性依次降低,所以铜、银、金的金属性越来越弱。
通过上述例子可以看出,玻恩-哈伯循环有助于理解不同金属活泼性不同的原因,并且根据其中具体数据还可以分析出哪种因素占主导。
4 共价型氢化物酸性的相对强度与玻恩-哈伯循环



表4 298K时HX(aq)电离过程的热力学数据
5 含氧酸盐的溶解性与玻恩-哈伯循环
含氧酸盐属于离子化合物,其溶解过程可用玻恩-哈伯循环表示如下:

由上述循环可知,溶解过程可认为是离子晶体中的正负离子首先克服离子间的引力,从晶格中解离下来成为气态离子,然后进入水中并与极性水分子结合成水合离子的过程。含氧酸盐的溶解情况与这一过程的有关,小于0,且绝对值越大,含氧酸盐越易溶解,反之难溶。
表5 几种含氧酸盐的及溶解性

表5 几种含氧酸盐的及溶解性
?
可见,玻恩-哈伯循环在含氧酸盐溶解过程中的使用,可以帮助学习者更好地理解含氧酸盐的溶解性规律。
通过以上讨论,可以看到玻恩-哈伯循环在无机化学中有非常广泛的应用,它除了可计算离子化合物的晶格能,原子的电子亲和能、质子亲和能外,还可以帮助学习者加深对无机化学中元素及其化合物的某些性质的理解。
[1]北京师范大学,华中师范大学,南京师范大学.无机化学.第4版.北京:高等教育出版社,2003
[2]何国方.泰安师专学报,1996(6):59
