带电对Ga7As7团簇基态结构的影响
2012-11-14杨建宋
杨建宋
(杭州师范大学理学院,浙江 杭州 310036)
带电对Ga7As7团簇基态结构的影响
杨建宋
(杭州师范大学理学院,浙江 杭州 310036)
采用全势能线性Muffin-tin轨道分子动力学计算方法(FP-LMTO-MD),对中性砷化镓Ga7As7团簇基态结构带电后在能量和几何结构上的变化进行研究.计算结果表明,随着电离程度的增加,团簇结构上的畸变更加明显,并且正离子团簇将比负离子团簇更快地失去稳定性.
砷化镓;团簇基态;结构畸变
0 引 言
砷化镓(GaAs)混合型团簇的能量与稳定结构,在近30年的研究中,已经有了一些成熟的理论计算方法和量子化学计算软件[1-8],尤其是原子个数在18以内并呈现砷原子和镓原子个数对称的团簇,其基态结构均已被找到[2].


图1 基于密度泛函理论计算下所得的Ga7As7团簇的基态结构(图中深色球表示砷原子,浅色球表示镓原子)Fig. 1 The ground-state structure of neutral Ga7As7 calculated by the DFT method

本文注意到了上述对Ga7As7团簇研究的进展,采用基于分子动力学原理的FP-LMTO-MD方法对Ga7As7团簇的已有结构和一些新构想的结构进行大量的优化计算,旨在寻找在FP-LMTO-MD方法计算下是否还存在具有更低能量的团簇结构,并进一步分析这个结构,研究其带电后在结构和稳定性方面随带电情况发生的演变.部分工作已经简要地发表在文献[21-22],这里将给出详细的计算数据和分析结果.
1 方 法
全势能线性Muffin-tin轨道分子动力学方法(FP-LMTO-MD)是一种在局域密度近似下求解Kohn-Sham方程的自洽迭代方法[23-26].在这种方法中,实空间被划分成两个部分:以原子核为中心的一个个非交叠的muffin-tin(MT)球和剩余的球间隙区.线性Muffin-tin轨道(LMTO)在MT球内取缀加的Hankel函数.在分子动力学计算过程中,经过几万到几十万次的迭代优化计算后,当总能量变化小于10-5a.u.和作用力小于10-3a.u.时,可认为它们已达到自洽收敛[27-30].
这个方法对研究团簇结构和能量是非常有效的,本课题组曾经用这种方法对中等以下Sin团簇和Gen团簇进行过广泛的研究,得到的结果与用其他先进的分子动力学方法计算所得到的相一致[31-32].为了比较,笔者把用这个方法对小硅团簇Si2~8计算所得到的标度后的结果和用MP4/6-31G*计算方法得到的结果及实验结果列于表1,可以看到有关数据能达到很好的一致性.
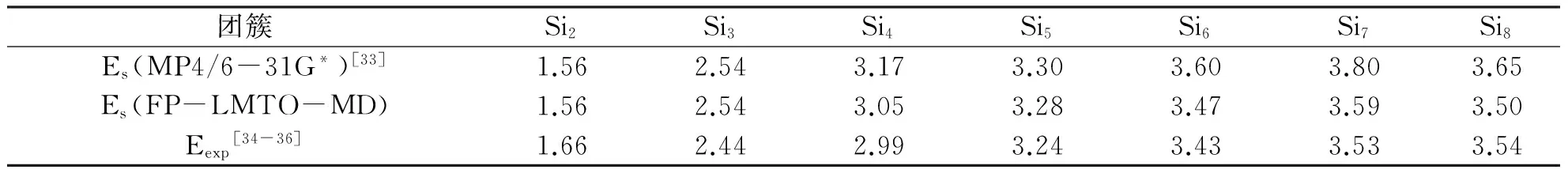
表1 计算所得的标度结合能及由Knudsen质谱仪测得的结合能Tab. 1 Calculated scaled cohesive energy and the measured cohesive energy by Knudsen mass spectrometers /eV
2 结构和讨论
2.1中性Ga7As7团簇的基态结构
笔者用FP-LMTO-MD方法对中性的Ga7As7团簇进行了优化计算.初始几何构形来自于两个方面:一是其他学者已经给出的低能稳定结构,如文献[2,18-20]中所给出的基于密度泛函理论得到的Ga7As7的基态结构;二是根据Ga6As6等更小的团簇稳定结构,依一定的对称性通过添加砷原子和镓原子重构的新结构.每一个初始几何构形,均用FP-LMTO-MD方法在计算机上进行无任何约束的几万次到几十万次的迭代计算.结果发现文献[2,18-20]中的Ga7As7基态结构并不是最低能量结构,笔者找到的Ga7As7团簇的最低能量结构是一个具有两个帽原子的三角棱柱结构,如图2中GaAs7a所示.该结构的总能量为-790 554.898 eV,比文献[2,18-20]中给出的“基态Ga7As7篮状五角棱柱结构”的总能量(-790 552.121 eV)低2.78 eV!在这个结构中,一个双帽镓原子处在三角棱柱的三角面上,另一个处在棱柱的矩形面上.中性的GaAs7a是一个具有两个价电子的满壳层电子结构,将是一个单态结构.在计算中笔者也发现一个与之前研究相同的现象,即在GaAs结构中,镓原子比砷原子更容易占据帽原子的位置.
2.2Ga7As7团簇的基态结构发生电离后的演变情况
团簇丢失或吸附一个或多个电子后将成为正或负离子团簇.在以往的研究中,发现离子团簇和中性团簇一样重要,在实验中离子团簇往往是伴随着中性团簇一起产生的.已有许多学者在开展中性团簇研究的同时也对离子团簇进行过研究.笔者对Ga7As7团簇基态结构(GaAs7a)发生电离后的演变情况进行了计算和分析,此时带电团簇中各原子坐标可参见表2,团簇的几何结构可参见图2.在这里,GaAs7a是中性团簇的基态结构,1+、2+和3+分别是其丢失1~3个电子后形成的正离子团簇,1-、2-、3-和4-分别是其获得1~4个电子后形成的负离子团簇.从图2中可以发现,让中性GaAs7a基态团簇离子化后,其稳定结构的几何形状将发生明显的变化.尤其是当两个电子或更多电子从中性团簇加上或离开后,团簇的几何结构相比于中性时将发生严重的畸变.

图2 Ga7As7团簇的基态结构在电离后的结构变化(图中深色球表示砷原子,浅色球表示镓原子)Fig. 2 Structural evolution of the ground state structure of neutral Ga7As7 clusters after the ionization
续表

结构原子xyz结构原子xyz3+AsAsAsAsGaGaGaGaAsAsAsGaGaGa1.4919916-2.0651631-0.76657391.33937514.0168028-4.48105320.0849574-1.1812572.0018418-1.0208642-1.74347821.6491575-0.58375151.39751222.32209842.3142163-1.188346-1.2774290.63897911.32477472.0558527-4.3844581.2725624-1.6710580.3397238-0.1375931.2938811-2.84036-1.8291230.6622551-1.4050240.0698809-1.3723851.6912131.97301132.67488850.42383481.13491662.4039876-2.72271-0.997059-2.352044-As1.58779351.9288927-2.296601As-1.79304552.52814390.7684783As-1.4476085-0.578885-1.23749As2.1089643-1.85716-0.411403Ga3.48007940.6773845-0.873062Ga-3.67252960.37447910.1670582Ga0.14685041.98253332.5263453Ga0.7199161-2.2091571.8979462As1.78611560.17869621.2325171As-1.7775458-1.5691731.0658821As-2.14266160.65828762.4805339Ga1.0174257-0.498583-2.716627Ga0.11167191.2453189-0.399025Ga0.0823124-2.736411-1.859435
注:坐标单位为10-10m.
从中性Ga7As7团簇的基态结构出发电离演变而成的各价正、负Ga7As7离子团簇,在结构畸变方面带负电的团簇会比带正电的团簇更重一些,尤其表现在一价的正、负离子团簇与中性团簇进行比较时.这个现象的物理原因可以通过它们的成键特性去理解:由于砷原子和镓原子的电子组态分别为4s24p3和4s24p1,砷原子可有sp,sp2,sp3或sp3d1杂化轨道,而镓原子仅有sp或sp2杂化轨道,这样就造成在砷化镓离子团簇中,镓原子比砷原子更容易丢失电子,而获得电子的可能性却比砷原子小.在它们各原子发生这种不对称的带电以后,存在于各带电原子间的静电斥力会使结构畸变增加.
计算结果也进一步表明,从同一中性团簇出发离子化演变后所获结构的稳定性,不仅与带电的数量有关,还与电离的电性有密切的联系.随着电子逐个从中性团簇中移去,离子团簇结构就迅速变得不稳定,通常当从中性结构出发减去3个或4个电子时,即使进行数百万次迭代计算,笔者还是没办法再找到其稳定结构.但当在中性团簇的基础上加上4个或5个电子时,即使其最终结构相比于中性结构来说有严重的畸变,团簇结构稳定性却仍然比较好.究其原因,还是来源于它们的成键特性.在正离子情况下,所谓电子丢失,实际上是镓原子在失去电子,随着电离程度的增加,砷原子与镓原子间的键长有明显的增加,即这种键的强度在迅速减弱,从而使正离子团簇的稳定性随电子数目的减少而迅速下降.
离子结构的HOMO-LUMO能隙列在表3中.可以看到大部分结构具有大于1 eV的能隙,正离子团簇1+和2+的能隙非常接近于其中性团簇结构的1.30 eV,负离子团簇1-和2-能隙只有中性时的3/4左右,原因是在负离子团簇中增加的一个电子将占据最低的未占有分子轨道(LUMO),它将明显地影响能隙Eg.3+和3-结构的能隙都非常小,表明这些结构将与团簇的中性结构有明显的结构畸变.

表3 Ga7As7团簇基态结构(GaAs7a)离子化后的HOMO-LUMO能隙Tab. 3 The HOMO-LUMO gaps of the ionic structures corresponding to the ground-state structure (GaAs7a)
3 结 论
用全势能线性糕模轨道分子动力学方法(FP-LMTO-MD)对Ga7As7团簇以及带电后其基态结构的演变情况作了详细的研究.在计算中发现了一个能量很低的稳定结构(GaAs7a),这个带帽棱柱状的基态结构其能量要比其他学者用Dmol等方法得到的篮状基态结构低2.78 eV.笔者对这个基态结构离子化后的演变情况作了详细分析,发现随着带电情况的加深,其结构畸变将会越来越明显,稳定性也会随之下降.畸变程度与丢失或吸附电子有关,也与带电数量的多少有关.负离子团簇的结构畸变相对更加重一些,但正离子团簇更明显地表现为其稳定性的下降.当这个基态团簇丢失4个电子时,无法再找到其稳定的离子团簇结构.
李宝兴教授在本论文研究工作中给予了悉心的指导和大力帮助,谨致谢意!
[1] Howes M J, Morgan D V. Gallium arsenide: materials, devices, and circuits[M]. New York :Wiley,1985:10-21.
[2] Karamanis P, Bégué D, Pouchan C.Abinitiofinite field (hyper) polarizability computations on stoichiometric gallium arsenide clusters GanAsn(n=2~9)[J]. J Chem Phys,2007,127(9):094706-094715.
[3] Al-Laham M A, Raghavachari K. Theoretical study of Ga4As4, Al4P4, and Mg4S4clusters[J]. J Chem Phys,1993,98(11):8770-8776.
[4] Vasiliev L, Ogut S, Chelikowsky J R.Abinitioabsorption spectra of gallium arsenide clusters[J]. Phys Rev B,1999,60(12):8477-8480.
[5] Lou L, Wang L, Chibante L P F,etal. Electronic structure of small GaAs clusters[J]. J Chem Phys,1991,94(12):8015-8020.
[6] Graves R M, Scuseria G E.Abinitiotheoretical study of small GaAs clusters[J]. J Chem Phys,1991,95:6602-6606.
[7] Andreoni W. III-V semiconductor microclusters: structures, stability, and melting[J]. Phys Rev B,1992,45(8):4203-4207.
[8] Zhao Wei, Cao Peilin, Li Baoxing,etal. Study of the stable structures of Ga4As4cluster using FP-LMTO MD method[J]. Phys Rev B,2000,62(24):17138-17143.
[9] Zhao Wei, Cao Peilin. Study of the stable structures of the Ga5As5cluster using the full-potential linear-muffin-tin-orbital molecular-dynamics method[J]. J Phys: Condens Matter,2002,14(1):33-44.
[10] Yi J Y. Atomic and electronic structures of small GaAs clusters[J]. Chem Phys Lett,2000,325:269-274.
[11] Yang Jiansong, Li Baoxing, Zhan Shichang. Study of GaAs cluster ions using FP-LMTO MD method[J]. Phys Lett A,2006,348:416-423.
[12] 杨建宋.带电对Ga5As5团簇能量和结构的影响[J]. 杭州师范大学学报:自然科学版,2009,8(6):432-438.
[13] O’Brien S C, Liu Y, Zhang Q,etal. Supersonic cluster beams of III-V semiconductors: GaxAsy[J]. J Chem Phys,1986,84(7):4074-4079.
[14] Zhang Q L, Liu Y, Curl R F,etal. Photo-dissociation of semiconductor positive cluster ions[J]. J Chem Phys,1988,88(3):1670-1677.
[15] Liu Y, Zhang Q L, Curl R F,etal. Photodetachment and photofragmentation studies of semiconductor cluster anions[J]. J Chem Phys,1986,85(12):7434-7441.
[16] Taylor T R, Gmez H, Asmis K R,etal. Photoelectron spectroscopy of GaX2ˉ, Ga2Xˉ, Ga2X2ˉ, and Ga2X3ˉ (X = P,As)[J]. J Chem Phys,2001,115(10):4620-4631.

[18] Zhao Jijun, Xie Ruihua, Zhou Xiaolan,etal. Formation of stable fullerenelike GanAsnclusters(6≤n≤9): gradient-corrected density-functional theory and a genetic global optimization approach[J]. Phys Rev B,2006,74(3):035319(1-5).
[19] Gutsev G L, Johnson E, Mochena M D,etal. The structure and energetics of (GaAs)n, (GaAs)n-, and (GaAs)n+(n=2~15)[J]. J Chem Phys,2008,128:144707(1-9).
[20] Gutsev G L, O’Neal R H Jr, Saha B C,etal. Optical properties of (GaAs)nClusters (n=2~16)[J]. J Phys Chem A,2008,112(43):10728-10735.
[21] Yang Jiansong,Tang Fan,Li Baoxing. Study of Ga7As7cluster and its ions[J]. NSNTAIJ,2008,2(2/3):77-82.
[22] Yang Jiansong, Li Baoxing. First-principles study of Ga7As7ionic cluster and influence of multi-charge on its structure[J]. Chin Phys B,2010,19(9):097103(1-9).
[23] Methfessel M, Schilfgaarde M V.Abinitiomolecular dynamics in the full-potential lmto method: derivation of a practical force theorem[J]. Int J Mod Phys B,1993,7:262-265.
[24] Methfessel M, Schilfgaarde M V. Derivation of force theorems in density-functional theory: application to the full-potential LMTO method[J]. Phys Rev B,1993,48:4937-4940.
[25] Methfessel M. Elastic constants and phonon frequencies of Si calculated by a fast full-potential linear-muffin-tin-orbital method[J]. Phys Rev B,1988,38(2):1537-1540.
[26] Methfessel M, Rodriguez C O, Anderson O K. Fast full-potential calculations with a converged basis of atom-centered linear muffin-tin orbitals: structural and dynamic properties of silicon[J]. Phys Rev B,1989,40(3):2009-2012.
[27] Kohn W, Sham L J. Self-consistent equations including exchange and correlation effects[J]. Phys Rev,1965,140:A1133-A1138.
[28] Anderson O K. Linear methods in band theory[J]. Phys Rev B,1975,12(8):3060-3085.
[29] Anderson O K, Woolley R G. Muffin-tin orbitals and molecular calculations: general formalism[J]. Mol Phys,1975,26(4):905-927.
[30] Springborg M, Anderson O K. Method for calculating the electronic structures of large molecules: helical polymers[J]. J chem Phys,1975,87(12):7125-7145.
[31] Li Baoxing, Cao Peilin. Structures of Gen clusters (n=3~10) and comparisons to Sinclusters[J]. Phys Rev B,2000,62(23):15788-15796.
[32] Li Baoxing, Cao Peilin, Zhan Shichang. Ground state structures of Sin(n=11~25) clusters[J]. Phys Lett A,2003,316(3/4):252-260.
[33] Raghavachari K. Theoretical studies on silicon clusters[J]. Phase Transit,1990(24/25/26):61-90.
[34] Bachels T, Schäfer R. Binding energies of neutral silicon clusters[J].Chem Phys Lett,2000,324(5/6):365-372.
[35] Schmude R W, Ran Q, Gingerich K A,etal. Atomization enthalpy and enthalpy of formation of gaseous Si2and Si3from mass spectrometric equilibrium measurements[J]. J Chem Phys,1995,102(6):2574-2579.
[36] Ran Q, Gingerich K A, Schmude R W. Atomization enthalpy and enthalpy of formation of gaseous Si4from mass spectrometric equilibrium measurements[J]. J Chem Phys,1993,99(10):7998-8004.
TheInfluencesofElectrificationontheGround-stateStructureofGa7As7Cluster
YANG Jian-song
(College of Science, Hangzhou Normal University, Hangzhou 310036, China)
Using full-muffin-tin-orbital molecular-dynamics (FP-LMTO-MD) method, the energy and geometry changes of the ground-state structure of Ga7As7cluster after charged were studied in detail. The results show that along with the increase of the degree of ionization, the cluster structure distortion becomes more obvious, and the positive ion cluster will lose its stability faster than negative ion cluster.
Ga7As7; ground-state cluster; structural distortion
2012-08-06
国家自然科学基金项目(11274084).
杨建宋(1957—),男,教授,主要从事凝聚态物理和理论物理研究.E-mail: yjs@hznu.edu.cn
10.3969/j.issn.1674-232X.2012.06.014
O561
A
1674-232X(2012)06-0544-07
