白额纵沟纽虫(Lineus alborostratus Takakura)的线粒体基因组*
2012-01-10徐冬丽陈海霞孙世春
徐冬丽,陈海霞,时 伟,孙世春
(中国海洋大学海洋生物多样性与进化研究所,山东青岛266003)
动物线粒体基因组通常为闭合的双链环形DNA分子,结构简单、母系遗传,基因成分相对稳定,被广泛应用于系统发育与进化研究[1-2]。迄今,GenBank中公布了2 200多种后生动物线粒体全基因组序列。在已记录的约1 275种[3]纽虫中,仅有Cephalothrix hongkongiensis[4]、Lineus viridis[5]和Paranemertes cf.peregrina[6]3种完成了线粒体基因组全序列测序。另外,Turbeville和Smith[7]报道了Cephalothrix rufifrons长度为10.1 kb的线粒体DNA连续片段,Chen et al.[6]测得了Cephalothrix sp.的线粒体DNA大部分序列(有一段预测长度约500 bp的非编码区未测出)。纽形动物线粒体基因组研究在确定纽形动物门在后生动物中的系统地位方面已经取得了有益结果(相关研究普遍支持纽形动物属于真体腔冠轮动物Lophotrochozoa)[4-5,7],但现有的数据远不能满足纽形动物门子类群的系统发育分析。白额纵沟纽虫(Lineus alborostratus Takakura,1898)(无针纲Anopla:异纽目Heteronemertea)是西北太平洋沿岸的一种常见纽虫,本文将报道其线粒体基因组全序列,并分析其碱基组成、基因排列、t RNA二级结构、蛋白质编码基因的密码子使用特征等。目的在于为基于线粒体基因组数据的纽形动物系统发育研究和纽形动物线粒体基因组进化研究积累数据。
1 材料与方法
1.1 基因组总DNA的提取
白额纵沟纽虫标本采自青岛薛家岛,使用D6032-01(OMEGA)试剂盒提取全基因组DNA,于-20℃保存。实验中所用DNA来自同一个体。
1.2 DNA扩增和测序
首先用文献报道的引物扩增cox1、cox3、rrnS-rrn L 3个DNA片段,然后依据得到的序列设计特异性引物继续扩增,引物序列见表1。PCR反应扩增体系为:模板总DNA 0.5μL,上下游引物(10μmol/L)各1μL,HSTMReaction Mix 12.5μL,Taq DNA酶(2.5 U/μL)0.25μL,加纯水补至25μL。PCR反应的循环参数为:95℃预变性5 min,然后95℃变性30 s,48~54℃退火50 s,72℃延伸2 min,循环35次,最后72℃延伸10 min。扩增的DNA片段用凝胶回收试剂盒(OMEGA)纯化,与PMD18-T载体(Takara)连接后克隆,产物以ABI 3730测序仪测序(北京华大基因)。
1.3 序列拼接、分析
将测序得到的片段序列用Codon Code Aligner软件进行拼接并人工检查,得到线粒体DNA全序列。利用t RNAScan-SE 1.21[10]网站服务器(http://lowelab.ucsc.edu/tRNAscan-SE/)搜索t RNA基因(得到21个)并预测其二级结构,用RNAStructure 4.5[11]推测剩余的tRNA基因结构,结合RNAfold[12]查找及预测其他特殊二级结构。利用NCBI的BLAST(http://www.ncbi.nlm.nih.gov/BLAST)比对及相邻基因的边界确定rrn L和rrnS基因。利用DNASIS[13]识别蛋白质编码基因的起始密码子和终止密码子,并通过和相近物种的线粒体基因同源序列的比对来确定蛋白编码基因。用DAMBE[14]和MEGA 4.0[15]软件计算序列的碱基组成、蛋白质编码基因的密码子组成及其使用频率。
白额纵沟纽虫线粒体基因组序列已提交到Gen-Bank,序列号为:JN234382。

表1 白额纵沟纽虫线粒体基因组PCR扩增引物Table 1 PCR primers used to amplify the complete mitochondrial genome of Lineus alborostratus
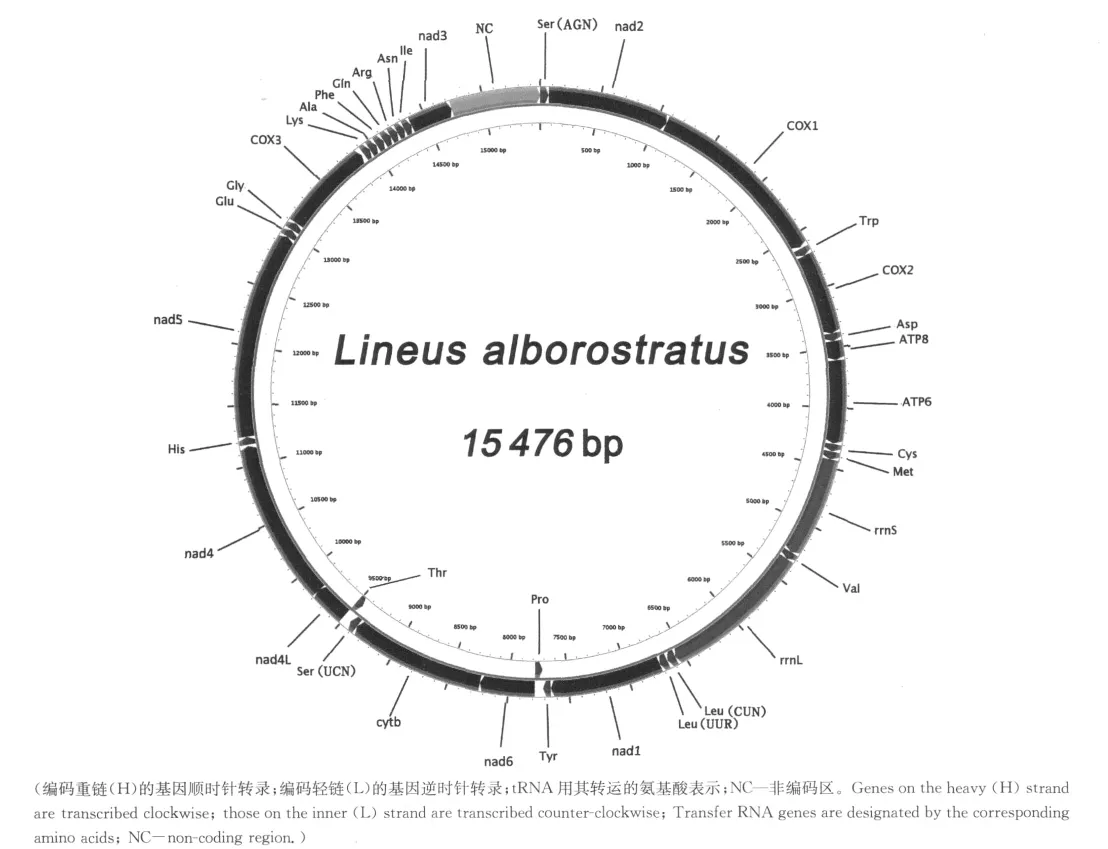
图1 白额纵沟纽虫线粒体DNA基因分布图Fig.1 Circular representation of the mtDNA of Lineus alborostratus.
2 结果与讨论
2.1 基因组构成及结构
白额纵沟纽虫线粒体基因组为闭合环状双链DNA,全长15 476 bp,与同属的L.viridis(15 388 bp)相近。具有后生动物线粒体基因组典型的基因组成,包括13个蛋白质编码基因(nad1-6,nad4L,cox1-3,atp6,atp8,cytb),22个t RNA基因,2个r RNA基因和1个主要的非编码区(见图1)。除tRNA-Thr和tRNA-Pro外,其他基因均编码在重链上。本种的基因排列顺序与L.viridis[5]一致,大部分基因长度差异较小,差异较大的是rrn L和非编码区,分别为1241bp/1305bp和747bp/415bp。基因紧密,除前述主要非编码区外只有总长72 bp的21处基因间隔。在cox2和trn D、nad4L和nad 4基因之间分别存在2和7 bp的重叠(见表2),类似碱基重叠也见于其他纽虫的线粒体基因组[4-6]。Boyce等[16]认为基因间的碱基重叠促进线粒体基因组小型化,而小型基因组复制所需时间短,具有一定的选择优势。

表2 白额纵沟纽虫线粒体基因组的基因分布Table 2 Location of genes in the mitochondrial genome of Lineus alborostratus
2.2 蛋白质编码基因
白额纵沟纽虫的蛋白质编码基因中,nad4L与nad 4基因间存在7个碱基的重叠。这2个基因的重叠亦见于C.hongkongiensis,C.sp和L.viridis[4-6],也常见于其他动物的线粒体基因组[17],可能nad4L/nad4间基因重叠具有一定的选择优势[16]。13个蛋白质编码基因均以ATG为起始密码子编码在重链上(见表2,图1)。除nad2、nad1和cytb基因使用不完全终止密码子“T”外,其余的10个蛋白质编码基因均以TAG或TAA终止。不完全终止密码子(T或TA)在动物线粒体DNA中常见[18],通常在转录后的加工过程中加接多聚腺苷酸尾,形成完整TAA终止密码子后终止翻译[1,19]。

图2 白额纵沟纽虫nad4基因上的发卡结构Fig.2 Hairpin structure in nad4 gene of Lineus alborostratus
所有蛋白质编码基因之间除nad4L/nad4,nad6/cytb,atp6/atp8和nad2/cox1基因外均以tRNA基因相连。这也是动物线粒体基因组的典型特征,可能有助于t RNA三叶草结构准确地将游离的氨基酸带到核糖体上的特定位点并添加到新生肽链的末端[19]。值得注意的是,白额纵沟纽虫的nad4基因(9 813~11 162 bp)上有一段37 bp的序列(10 052~10 088 bp)可以形成发卡结构(见图2),其AT含量高达81.1%。蛋白质编码基因上的茎环结构虽然也出现在其他纽虫线粒体中,一般都出现在2个基因连接的地方,并被认为是与多顺反子的启动或转录有关[6],这种出现在蛋白质编码基因内部的二级结构是否具有重要生物功能有待研究。
2.3 碱基组成和密码子使用
碱基组成的偏好性常通过偏倚度(Skewness)来描述,该指标能够衡量A对T和G对C的相对数量,分别以(A%-T%)/(A%+T%)和(G%-C%)/(G%+C%)来计算[20]。白额纵沟纽虫线粒体基因组各碱基百分含量为:T(41.2%)>A(23.5%)>G(22.8%)>C(12.5%)(见表3),重链(H链)呈现出较强的T偏好(SkewAT=-0.27)和较弱的C偏好(SkewGC=0.29)。13个蛋白质编码基因的A+T含量为62.9%,略低于全序列的A+T含量(64.7%),这种AT偏向性与其他纽虫相似[4-7]。密码子第三位点的A+T含量高于第一位点和第二位点(见表3),反映了该位点在进化过程中的选择压力较低,具有较高突变率[21]。所有基因中rrn L的A+T碱基含量最高(73.2%)。

表3 白额纵沟纽虫线粒体基因组碱基组成Table 3 Base compositions of the mitochondrial genome of Lineus alborostratus
白额纵沟纽虫线粒体基因组的密码子使用情况见表4,13个蛋白质编码基因共包含3 698个密码子(终止子除外)。氨基酸出现频率最高的是Leu(14.85%),其次为Phe(10.86%),两者都属于疏水氨基酸,这与线粒体基因组编码的大部分蛋白质是跨膜蛋白相适应。蛋白质编码基因偏好使用富含T、G和A的密码子,富含T的密码子(三联体密码子中至少含有2个T)出现1 570次,占42.35%,富含G、A、C的密码子(三联体密码子中至少含有2个G、A、C)出现频率分别为12.99%、10.94%和4.94%。对于多数氨基酸,以T结尾的密码子的出现频率明显高于其他同义密码子,如Phe(TTT,9.92%;TTC,0.94%)、Ile(ATT,4.96%;ATC,0.49%)(见表4)。同义密码子的偏好性被认为与沉默位点的选择有关,并且能够增强转录效率[22-23]。由于同义密码子并不改变最终的蛋白产物,所以对于那些频繁被使用的密码子的选择性被认为是很弱的[24]。

表4 白额纵沟纽虫线粒体基因组中蛋白质编码基因的密码子使用Table 4 Codon usage of protein-coding genes in the mitochondrial genome of Lineus alborostratus
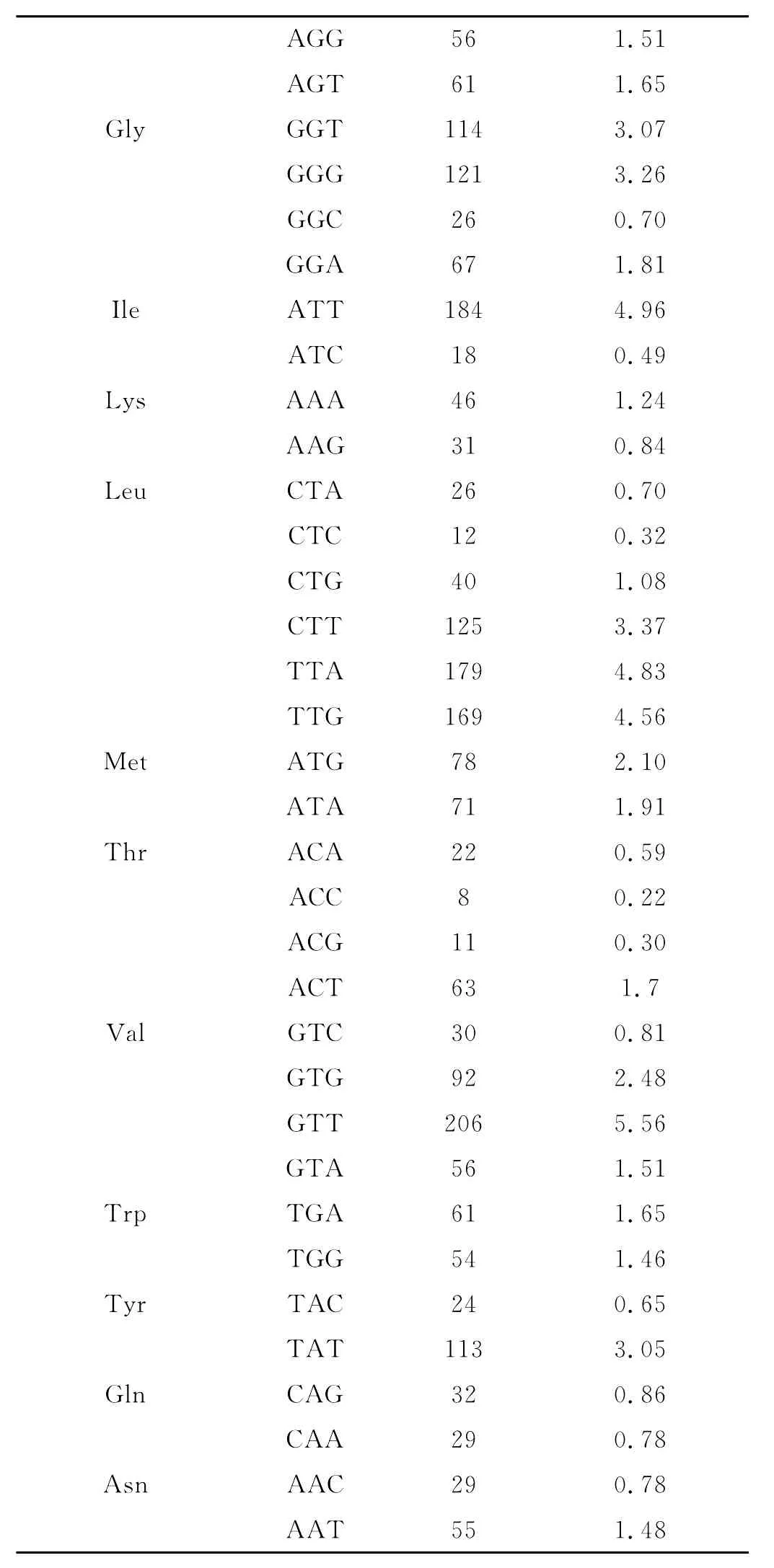
*终止密码子Stop codons
2.4 转运RNA和核糖体RNA
白额纵沟纽虫线粒体基因组含有22个tRNA,长度范围为62~71 bp,通过t RNAScan-SE 1.21只找到21个t RNAs,另外的tRNA-Arg基因根据反密码子,利用RNA structure 4.5软件辅助人工预测得到(见图3)。线粒体t RNA二级结构中除正常的碱基配对外,还存在4处碱基错配,分别发生在2个tRNA-Leu的氨基酸接受臂和tRNA-Gln、tRNA-Trp的TΨC臂上。这种t RNA上的错配碱基在其他纽虫线粒体基因组中均有发现[4-7],这些错配可以通过转录后RNA编辑来修正,并不会影响t RNA对相应氨基酸的转运功能[25-27]。另外还有大量的G-T配对,这在tRNA中是一种常见情况。

图3 白额纵沟纽虫线粒体基因组中预测的22个tRNA的二级结构。Fig.3 Secondary structures predicted for 22 tRNAs in the mitochondrial genome of Lineus alborostratus
在其他纽虫中有报道过缺失DHU臂的情况[5-6],在白额纵沟纽虫的tRNA中没有出现。值得注意的是,tRNA-Cys的TΨC环只有2个T碱基,这2个T碱基可能就是欠完整的TΨC环。
白额纵沟纽虫22个t RNA基因的反密码子与L.viridis线粒体t RNA的反密码子完全相同。与细首属(Cephalothrix)的3种纽虫及P.cf.peregrina相比,只有tRNA-Ser(AGN)的反密码子有异。tRNA-Ser(AGN)的反密码子在白额纵沟纽虫和L.viridis中为TCT,在其它纽虫中为GCT。
白额纵沟纽虫线粒体基因组中含有2个r RNA基因(rrnS和rrn L),其长度分别为835和1241 bp,以同一方向编码在重链上,并由tRNA-Val分隔开(见图1)。其中rrnS长度大于P.cf.peregrina(776 bp),与C.hongkongiensis、C.sp.和L.viridis(分别为838、836、833 bp)接近,rrn L的长度大于P.cf.pere-grina(1 093 bp),小于C.hongkongiensis、C.sp和L.viridis(分别为1 291,1 290和1 305 bp)[4-7]。
2.5 非编码区
基因nad3和t RNA-Ser(AGN)之间的有一段长度为747 bp的非编码区(non-coding region,NC),占基因组全长的4.83%,其AT含量为65.2%。在已测序纽虫的线粒体DNA中均发现了类似的非编码区,其中L.viridis非编码区的位置与本种一致[5],C.hongkongiensis和C.sp的主要非编码区位于tRNA-Tyr和tRNA-Cys之间(除主要非编码区外,这2种纽虫还具另外2个大于100 bp的非编码区)[4,6],P.perigrina则在nad 4L/nad4和tRNA-Trp/tRNASer(AGN)之间分别具有长度为101和114 bp的2段非编码区[6]。主要非编码区的长度在几种纽虫间变化很大,从114 bp(P.perigrina)到885 bp(C.hongkongiensis)[4-6]。与同属的L.viridis相比,白额纵沟纽虫长出332 bp(747 bp对415 bp)[5]。由于该区域所受的选择压力较小,与编码区相比具有更高水平的长度和位点多态性,不同动物线粒体基因组DNA大小的差异主要是由这个区域长度变化造成的[28]。
非编码区在已报道的纽虫都存在茎环结构[4-6],这类特殊二级结构在大多数后生动物中被认为是线粒体轻链复制起始位点[29-31]。但白额纵沟纽虫非编码区没有检测到类似结构。纽虫线粒体DNA非编码区AT含量普遍高于基因组AT含量,如在C.hongkongiensis为83.8%[4],在C.sp.一段389 bp的序列(非编码区未完全测出)中更高达92.5%[6]。因此,此非编码区也称为AT富含区[32]。白额纵沟纽虫非编码区65.2%的AT含量在已测纽虫中是最低的。此外,在白额纵沟纽虫的非编码区还发现了一些短的重复单元,如ATTTTTTA、AAAAAAT各有3个重复,GGGGG有4个重复。一般来说线粒体重复单元长度一般由1~数百bp不等,重复数也可以从2个到几百不等,其产生机制一般认为是由于链的滑动错配所导致[33-34]。
除了这个主要的非编码区外,白额纵沟纽虫线粒体基因组中还存在21处长度为1~12 bp的核苷酸插入区,总长72 bp,2处12 bp间隔分别位于cox1和tRNA-Trp、nad4和tRNA-His之间。相比L.viridis总长248 bp的24处核苷酸插入[5],白额纵沟纽虫的插入核苷酸明显较少。
[1] Anderson S,Bankier A T,Barrell B G,et al.Sequence and organization of the human mitochondrial genome[J].Nature,1981,290(5806):457-465.
[2] Nishibori M,Shimogiri T,Hayashi T,et al.Molecular evidence for hybridization of species in the genus Gallus except for Gallus varius[J].Anim Genet,2005,36(5):367-375.
[3] Kajihara H,Chernyshev A V,Sun S C,et al.Checklist of nemertean genera and species published between 1995 and 2007[J].Spec Diver,2008,13(4):245-274.
[4] Chen H X,Sundberg P,Norenburg J L,et al.The complete mitochondrial genome of Cephalothrix simula(Iwata)(Nemertea:Palaeonemertea)[J].Gene,2009,442(1-2):8-17.
[5] Podsiadlowski L,Braband A,Struck T H,et al.Phylogeny and mitochondrial gene order variation in Lophotrochozoa in the light of new mitogenomic data from Nemertea[J].BMC Genomics,2009,10:364.doi:10.1186/1471-2164-10-364.
[6] Chen H X,Sundberg P,Wu H Y,et al.The mitochondrial genomes of two nemerteans,Cephalothrix sp.(Nemertea:Palaeonemertea)and Paranemertes cf.peregrina(Nemertea:Hoplonemertea)[J].Mol Biol Rep,2011,38:4509-4525.
[7] Turbeville J M,Smith D M.The partial mitochondrial genome of the Cephalothrix rufifrons(Nemertea,Palaeonemertea):charac-terization and implications for the phylogenetic position of Nemertea[J].Mol Phylogenet Evol,2007,43(3):1056-1065.
[8] Folmer O,Black M,Hoeh W,et al.DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates[J].Mol Mar Biol Biotechnol,1994,3(5):294-299.
[9] Boore J L,Brown W M.Mitochondrial genomes of Galathealinum,Helobdella,and Platynereis:sequence and gene arrangement comparisons indicate that Pogonophora is not a phylum and Annelida and Arthropoda are not sister taxa[J].Mol Biol Evol,2000,17(1):87-106.
[10] Lowe T M,Eddy S R.tRNAscan-SE:a program for improved detection of transfer RNA genes in genomic sequence[J].Nucleic Acids Res,1997,25(5):955-964.
[11] Reuter J S,Mathews D H.RNAstructure:software for RNA secondary structure prediction and analysis[J].BMC Bioinformatics,2010,11:129.doi:10.1186/1471-2105-11-129.
[12] Denman R B.Using RNAFOLD to predict the activity of small catalytic RNAs[J].Biotechniques,1993,15(6):1090-1095.
[13] Mandler J,Ludwig S.SUBSIS:DNASIS/PROSIS subdirectory management[J].Comput Appl Biosci,1990,6(3):291-292.
[14] Xia X,Xie Z.DAMBE:software package for data analysis in molecular biology and evolution[J].J Hered,2001,92(4):371-373.
[15] Tamura K,Dudley J,Nei M,et al.MEGA4:Molecular Evolutionary Genetics Analysis(MEGA)software version 4.0[J].Mol Biol Evol,2007,24(8):1596-1599.
[16] Boyce T M,Zwick M E,Aquadro C F.Mitochondrial DNA in the bark weevils:size,structure and heteroplasmy[J].Genetics,1989,123(4):825-836.
[17] Boore J L.Animal mitochondrial genomes[J].Nucleic Acids Res,1999,27(8):1767-1780.
[18] Clary D O,Wolstenholme D R.The mitochondrial DNA molecular of Drosophila yakuba:nucleotide sequence,gene organization,and genetic code[J].J Mol Evol,1985,22(3):252-271.
[19] Ojala D,Montoya J,Attardi G.tRNA punctuation model of RNA processing in human mitochondria[J].Nature,1981,290(5806):470-474.
[20] Perna N T,Kocher T D.Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes[J].J Mol Evol,1995,41(3):353-358.
[21] Plaisance L,Huyse T,Littlewood D T,et al.The complete mitochondrial DNA sequence of the monogenean Gyrodactylus thymalli(Platyhelminthes:Monogenea),a parasite of grayling(Thymallus thymallus)[J].Mol Biochem Parasitol,2007,154(2):190-194.
[22] Sharp P M,Matassi G.Codon usage and genome evolution[J].Curr Opin Genet Dev,1994,4(6):851-860.
[23] Duret L,Mouchiroud D.Expression pattern and,surprisingly,gene length shape codon usage in Caenorhabditis,Drosophila,and Arabidopsis[J].Proc Natl Acad Sci U S A,1999,96(8):4482-4487.
[24] Dey S.Benefits of being biased![J].J Genet,2004,83(2):113-115.
[25] Lavrov D V,Brown W M,Boore J L.A novel type of RNA editing occurs in the mitochondrial tRNAs of the centipede Lithobius forficatus[J].Proc Natl Acad Sci U S A,2000,97(25):13738-13742.
[26] Yokobori S,Paabo S.Transfer RNA editing in land snail mitochondria[J].Proc Natl Acad Sci U S A,1995,92(22):10432-10435.
[27] Yokobori S,Paabo S.Polyadenylation creates the discriminator nucleotide of chicken mitochondrial tRNA(Tyr)[J].J Mol Biol,1997,265(2):95-99.
[28] Wolstenholme D R.Animal mitochondrial DNA:structure and evolution[J].Int Rev Cytol,1992,141:173-216.
[29] Pegoraro L,Yang Z,Samake S,et al.Sequence comparison of mitochondrial tRNA genes and origin of light strand replication in Bos taurus and Nellore(Bos indicus)breeds[J].Anim Genet,1996,27(2):91-94.
[30] Shadel G S,Clayton D A.Mitochondrial DNA maintenance in vertebrates[J].Annu Rev Biochem,1997,66:409-435.
[31] Macey J R,Larson A,Ananjeva N B,et al.Evolutionary shifts in three major structural features of the mitochondrial genome among iguanian lizards[J].J Mol Evol,1997,44(6):660-674.
[32] Zhang D X,Hewitt D M.Insect mitochondrial control region:a review of its structure,evolution and usefulness in evolutionary studies[J].Biochem Syst Evol,1997,25:99-120.
[33] Buroker N E,Brown J R,Gilbert T A,et al.Length heteroplasmy of sturgeon mitochondrial DNA:an illegitimate elongation model[J].Genetics,1990,124(1):157-163.
[34] Faber J E,Stepien C A.Tandemly repeated sequences in the mitochondrial DNA control region and phylogeography of the pikeperches Stizostedion[J].Mol Phylogenet Evol,1998,10(3):310-322.
