正十二烷高温燃烧机理的构建及模拟
2011-12-11华晓筱王静波王全德谈宁馨李象远
华晓筱 王静波 王全德 谈宁馨,* 李象远
(1四川大学化学工程学院,成都610065; 2四川大学化学学院,成都610064)
正十二烷高温燃烧机理的构建及模拟
华晓筱1王静波1王全德2谈宁馨1,*李象远1
(1四川大学化学工程学院,成都610065;2四川大学化学学院,成都610064)
基于燃料燃烧反应机理的计算机自动生成方法,构建了正十二烷高温燃烧的详细反应机理;分别采用物质产率分析和反应路径流量分析方法对详细机理进行简化,得到包含202个物种、738步反应的半详细机理和53个物种、228步反应的骨架机理;对正十二烷点火延时、高温裂解以及层流火焰速度的模拟结果表明半详细机理和骨架机理具有很高的模拟精度,在工程计算流体力学仿真设计中有很好的应用前景.最后分析了正十二烷高温燃烧的反应路径,并对点火延时做了敏感度分析,考查了机理中的关键反应.
正十二烷;燃烧机理;机理简化;化学动力学模拟
1 引言
随着计算流体力学以及计算燃烧学的不断发展,燃烧数值模拟在工程设计中扮演着越来越重要的角色.精确模拟航空燃料燃烧的化学动力学过程对于航空发动机的设计具有重要的指导作用.然而,航空煤油是由多种高碳烃化合物所组成,其组分比较复杂,直接模拟它的化学动力学过程相当困难,目前通用的方法就是选择航空煤油中几种代表性的碳氢化合物组成替代燃料,来模拟实际燃料的物理化学性质.1-3正十二烷是航空煤油替代组分的常用化合物,构建正十二烷合理的燃烧机理对于研究航空燃料的燃烧动力学具有重要意义.
燃料燃烧的反应机理研究在国外已开展了二十余年.3对于直链烷烃的机理,已有诸多研究.4-7如Westbrook等4依据反应类规则,构建了C8-C16直链烷烃的高温和低温燃烧机理(2116个物种,8130步反应).Wang等5构建了用于直链烷烃(包括C7-C12)的高温燃烧机理JetSurf(194个物种,1458步反应),该机理在直链烷烃的高温段的数值模拟结果与实验值吻合得很好,但两者都不是针对正十二烷燃烧的专用机理;Wang研究小组根据链烃高温燃烧的特性,直接将正十二烷裂解成小分子片段,并结合核心机理,构建了适用于正十二烷高温燃烧的机理(171个物种,1306步反应).2这些机理的物种数和反应数较多,且未发现经过简化的正十二烷的燃烧机理,难以在工程计算流体力学模拟中直接应用;在国内,还没有正十二烷燃烧机理的构建研究.
本课题组拟以正十二烷的高温燃烧机理的构建为例,意在初步探索适用于计算流体力学的高碳烃燃烧反应简化机理的构建方法.为此,在本课题组首先开发的反应机理自动生成程序ReaxGen8的基础上,构建了正十二烷的高温燃烧详细机理,并对详细机理进行了简化和模拟验证.
2 反应机理构建
2.1 详细机理构建
机理自动生成程序ReaxGen依据如下原理产生反应机理:烷烃的高温燃烧反应以自由基的链反应机理为主要特征;高温燃烧反应的种类有限,具备程序化的特征;假定大于C4的物种参与反应时,反应中心受分子大小和周围环境的影响很小,其反应动力学参数按反应归类的方式确定.9
链烷烃的高温燃烧反应类型主要有:4,6(1)烷烃的单分子裂解反应,(2)自由基对烷烃的氢提取反应,(3)烷基的裂解反应,(4)烷基的氢迁移重排,(5)烷基氧化生成烯烃,(6)烯烃自由基的裂解反应,(7)烯烃的裂解反应,(8)烯烃烯丙基、烯基、烷基位氢提取反应,(9)氢原子、氧原子、甲基、羟基、过氧化氢自由基的烯烃加成,(10)逆烯反应,(11)烷基位烯自由基的重排反应.
根据以上反应类型构建相应的子程序模块,高碳烃的起始反应物调用相应的反应方程,所得的产物也调用对应的反应方程,由此就产生一连串的链反应及相应的动力学参数,再结合碳原子数小于等于4的物种构成的核心机理(本文采用Wang等10发展的USCMechⅡ机理),得到高碳烃高温燃烧反应的动力学文件.
除核心机理外,物种的热力学数据根据基团贡献法计算;C0
-C4的高温核心反应的热力学数据来源于文献.10物种的输运数据由物质的临界参数计算,11对于缺乏临界参数的自由基等物质,采用与其结构最接近的中性分子的数据近似.
由Reaxgen自动生成的正十二烷的详细反应机理,包含1008个物种和4105步反应;为了提高计算效率,首先用物质产率分析方法简化机理,此方法先计算每个反应在总的反应过程中,对每个物种的生成或消耗的贡献,如果贡献小于某一阈值,则删除该反应,最后把没有参与最终机理任何反应的多余物种删掉,由此得到的半详细机理包含202个物种,738步反应.
2.2 骨架机理的构建
基于最近发展的反应路径流量分析(PFA)方法,12编写了与Chemkin-SENKIN13,14接口的简化程序,采用Lu和Law15提出的多步简化方法,简化了正十二烷的半详细反应机理.PFA方法是在直接关系图(DRG)的基础上提出来的.DRG方法的基本思路为:如果机理中某组分B的去除导致组分A的生成或消耗产生较大误差,则对于组分A来说,组分B是重要的.在DRG中定义如下关系:15,16

式(1)中,rAB为组分B对A的正规化贡献,ωi为第i个基元反应的净反应速率,νA,i为组分A在第i个基元反应的化学计量系数.可以看到,rAB足够大时,去除组分B将导致组分A的生成或者消耗产生较大误差.因此,组分A依赖于组分B.具体编程实现时,初始设定一个阈值ε,选定初始重要组分,采用深度优先搜索方法等进行搜索,然后删除不重要的物种及其涉及的反应,得到简化的骨架机理.PFA方法的思想与DRG类似,区别在于计算rAB时,把物种B对A的生成和消耗进行分离,其定义如下式:12
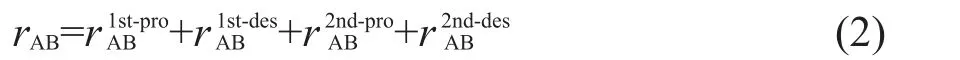
上式中,r1st-proAB和r1st-desAB定义如下:

式(3)和(4)中,PAB和DAB分别代表组分B的存在对组分A的生成和消耗的影响,PA和DA分别代表组分A的生成量和消耗量.考虑组分A和组分B通过其他物质M之间的间接相互影响,PFA方法中定义了如下关系表示组分A和组分B之间的二级相互作用:

为了简化,组分B对A的影响采用式(2)的加和形式表示.已有的机理简化结果表明,PFA方法能够有效地减少详细机理的物种数目,同时保留主要的反应路径.12,17
为了最大限度地简化详细机理同时保留详细机理的合理性,本文采用多步简化方法.15为了使得到的骨架机理具有比较宽的应用范围,本文对Chemkin-SENKIN在温度范围为1000-1800 K,压力从1.0×105、5.0×105Pa到3.0×106Pa和当量比(燃料-氧气实际燃烧的体积比除以燃料-氧气燃烧的化学恰当体积比)为0.5、1.0、1.5的点火模拟过程进行反应速率抽样,选择正十二烷、氧气和氮气为初始组分,对不同阈值条件下得到的骨架机理在抽样范围内的点火延时模拟结果与半详细机理进行比较,在可接受的误差内最终得到的骨架机理包含53个物质,涉及到228步反应.图1给出了骨架机理与半详细机理在不同当量比和压力下的点火延时模拟结果.点火延时τign定义为从反应起始时刻到OH浓度随时间的变化率达到最大((dcOH/dt)|max)的时刻之间的间隔.式中cOH为OH的浓度,t为反应时间.由图1可以看到,骨架机理模拟结果的变化趋势在较宽的模拟范围内与半详细机理的模拟结果能较好吻合,验证了简化模型的合理性.
3 半详细机理以及骨架机理的模拟验证
3.1 正十二烷的高温裂解模拟
You等2的研究结果表明,高温条件下,直链烷烃的裂解反应非常快,只要能正确地预测裂解产物的分布,加上小分子化合物的燃烧核心机理,就能有效地预测直链烷烃在高温燃烧下的点火延时、火焰传播速度等.因此,为了验证机理能否有效地预测正十二烷的裂解产物分布,本文首先模拟了高温条件下正十二烷裂解转化率及裂解产物分布的情况,并与实验结果18比较.采用Chemkin-II程序包中的全混流反应器模型,模拟了正十二烷(摩尔分数为2%)在氦气中的裂解.在滞留时间为1 s,压力为1.0× 105Pa,温度在800-1100 K范围的条件下,正十二烷的转化率以及主要产物甲烷、乙烯随温度的变化分布结果见图2.

图1 半详细机理与骨架机理的点火延时模拟结果Fig.1 Predicted ignition delay time by using the detailed and reduced mechanismsϕ:equivalence ratio,τign:ignition delay time

图2 正十二烷高温裂解结果Fig.2 High-temperature pyrolysis results of n-dodecane
半详细机理及骨架机理的模拟结果均很好地预测了正十二烷高温裂解的转化率以及主要产物的分布.其中,半详细机理转化率的模拟结果与实验值吻合得很好,且准确地预测了乙烯(C2H4)的生成,但是CH4的模拟结果略微偏高.由于骨架机理的抽样来自燃烧模拟过程,骨架机理的误差有所增加,其模拟结果比半详细机理的结果小.尽管如此,骨架机理仍然较好地预测了燃料的转化率和主要产物的分布,其中CH4的模拟结果与实验值能很好吻合.分析图中结果可知,随着温度升高,正十二烷转化率增大,小分子产物增多,其中乙烯最多,C―C键的断裂在裂解中起着越来越重要的作用.
3.2 点火延时
本文采用反射激波管模型模拟了正十二烷在不同条件下的点火延时.实验方面,Vasu等19用反射激波管研究了正十二烷在727-1422 K,1.5×106-3.4×106Pa,当量比为0.5和1.0条件下燃烧的点火延时;Shen等20也研究了正十二烷在较高压力下燃烧的点火延时特性.本文用半详细机理和骨架机理模拟了当量比为0.5,压力分别为1.4×106和2.0×106Pa时,正十二烷高温燃烧的点火延时,并与实验值比较.同时,为了更好地验证机理,选用国际上已公开的JetSurf机理5进行了对比模拟,结果如图3所示.本文的机理模拟结果比JetSurf模拟结果更接近实验值,且温度越高,与实验值吻合得越好.由于本文主要研究正十二烷的高温燃烧机理,因此随着温度降低至直链烷烃燃烧的负温度区域,点火延时模拟的误差开始增大,因此需要进一步开展低温燃烧机理的研究.同时,本文的半详细机理和骨架机理在其他当量比下的模拟结果也比较好,可见构建的半详细机理和骨架机理能很好地预测正十二烷高温点火的特性.
3.3 层流火焰速度

图3 正十二烷在不同压力下的点火延时结果Fig.3 Ignition delay time of n-dodecane at different pressures
采用PREMIX程序21结合Chemkin 2.0模拟了正十二烷在不同当量比条件下的层流火焰速度,模拟过程中采用混合平均方法求解输运数据.22图4给出了正十二烷在初始温度为403 K,压力为1.0×105Pa时,层流火焰速度随当量比的变化,并与Ji23和 Kumar24等的实验结果比较.同时也采用了JetSurf机理进行模拟,以便于比较.图4表明本文的半详细机理和骨架机理和文献23的实验值吻合得很好.在当量比大于1时,文献24给出的火焰速度与文献23和半详细机理及骨架机理模拟的结果相差较大,是因为文献24的数据是通过线性外推得到的,而文献23的数据由非线性外推得到.

图4 初始温度为403 K时正十二烷的层流火焰速度随当量比(ϕ)变化的模拟结果Fig.4 Laminar flame speed of n-dodecane versus equivalence ratio(ϕ)at fresh gas temperature 403 K
4 反应路径分析
反应路径分析对于认识不同燃料燃烧的动力学过程,进一步优化反应机理具有重要意义,反应路径分析是考虑不同的反应m(m=1,2,…,M)对化学组分n(n=1,2,…,N)的形成(或消耗)的贡献的百分数,对于以时间为变量的均相燃烧系统,如激波管模型,为了从整体上描述正十二烷的燃烧反应路径,需要对时间积分,才能获得整个燃烧过程中主要组分的生成或消耗的整体反应路径.由于采用反应路径流量分析方法得到的骨架机理能较好地保留主要的反应通道,因此本文采用骨架机理进行路径分析.图5给出了在1.0×105Pa,1149 K和当量比为0.5的反射激波管模型模拟的反应路径.
由图可见,由C―C键直接断裂产生的1-辛基和1-壬基各占0.1%,而由H、O、OH对正十二烷进行氢提取反应,生成六种十二烷基的反应占99.8%,这表明在温度较高的情况下,正十二烷的初始反应主要是小分子自由基在正十二烷的不同碳原子部位进行氢提取反应,产生六种十二烷基;这六种十二烷基可以快速地发生异构反应.也可进一步通过β-断裂反应,生成小分子的烯烃和较小的烷基,同样,这些小分子烷基继续发生异构化反应,然后继续通过β-断裂反应生成更小的烯烃和烷基.综合分析6种十二烷基的异构化情况,可知主要生成2-十二烷基,它首先经过β-断裂形成丙烯(C3H6)和1-壬基(1-C9H19);1-C9H19可以直接去氢,生成壬烯,但是这步反应相对比较慢,1-C9H19主要还是通过快速的异构化反应生成5-C9H19等,然后经由β-断裂,最终得到小分子碳氢物质(如甲烷、乙烯、甲基、乙基、1,3-丁二烯等).
5 敏感度分析
敏感度分析方法对于认识燃料燃烧动力学中的关键反应以及机理简化等具有重要作用.为了考查正十二烷燃烧反应中,对点火延迟影响较大的关键反应,本文采用文献25所用的方法,对正十二烷点火延时做敏感度分析.具体方法为:首先,选择OH作为关键物质,用ChemKin软件在点火模拟过程中作敏感度分析,根据敏感度系数的大小对所有反应进行排序;然后,挑选敏感度系数较大的反应,这些反应的动力学参数的指前因子乘以2,即整个反应速率ki增加一倍;进一步模拟点火延时,计算点火延时的变化率:

图5 正十二烷高温燃烧反应路径图Fig.5 Reaction path analysis for high-temperature combustion of n-dodecane

图6 各反应的速率对点火延时的敏感度分析Fig.6 Sensitivity analysis of ignition delay time with respect to the reaction rate
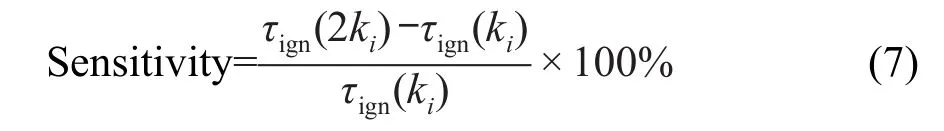
反应的敏感度系数为正,说明此反应对点火起抑制作用,若为负则起促进作用.图6给出了1.0×105Pa,当量比为0.5,温度1149 K点火模拟条件下的敏感度分析的结果.
由图6可见,反应R1、R3、R4、R5、R6、R7均对点火起促进作用,其中链反应R1(H+O2=O+OH)对点火延迟的促进作用影响最大;R2、R8-R12对点火有抑制作用,其中R2(C2H3+O2=CHO+HCHO)对点火的抑制作用最大.由图6可见,核心机理中的关键小分子或自由基(如HO2、H、OH)的反应对点火影响很大,而扩展机理中(碳原子数大于4的物种所构成的机理)的大分子及自由基的反应对点火的影响较小,只有R15、R16能观察到明显的影响.
6 结论
本文运用燃烧化学动力学的计算机自动生成程序Reaxgen,构建了正十二烷的高温燃烧详细机理,先由物质产率分析方法简化得到半详细机理,再由反应路径流量分析方法简化得到骨架机理.为验证机理的合理性,模拟了正十二烷的高温裂解、高温燃烧的点火延时和层流火焰速度;结果表明:半详细机理及骨架机理均能很好地反映正十二烷的裂解转化率及小分子产物的分布情况,模拟得到的点火延时、层流火焰速度均与实验值吻合较好.分析正十二烷的高温燃烧的反应路径,发现小分子自由基在不同部位对正十二烷进行氢提取反应,产生六种十二烷基;这些烷基可以快速地发生异构反应,也可以进一步通过β-断裂反应,生成小分子的烯烃和较小的烷基,进入核心机理;通过敏感度分析方法,找出了对点火延时促进最大的反应H+O2=O+ OH和抑制最大的反应C2H3+O2=CHO+HCHO.
本文以正十二烷的高温燃烧机理的构建为例,说明了基于计算机软件辅助机理生成,有助于快速全面地生成复杂燃料的详细燃烧机理,再结合自动化的机理骨架构筑方法,得到的高碳烃化合物燃烧反应机理,与人工拟定的方法相比,效率更高,该方法在构建航空煤油等复杂燃料中各组分的燃烧机理时具有广泛的应用前景.
(1) Dagaut,P.;Cathonnet,M.Prog.Energy Combust.Sci.2006,32, 48.
(2)You,X.Q.;Egolfopoulos,F.N.;Wang,H.Proc.Combust.Inst. 2009,32,403.
(3) Simmie,J.M.Prog.Energy Combust.Sci.2003,29,599.
(4) Westbrook,C.K.;Pitz,W.J.;Herbinet,O.;Curran,H.J.;Silke, E.J.Combust.Flame 2009,156,181.
(5) Sirjean,B.;Dames,E.;Sheen,D.A.;You,X.Q.;Sung,C.; Holley,A.T.;Egolfopoulos,F.N.;Wang,H.;Vasu,S.S.; Davidson,D.F.;Hanson,R.K.;Bowman,C.T.;Kelley,A.; Law,C.K.;Tsang,W.;Cernansky,N.P.;Miller,D.L.;Violi,A.; Lindstedt,R.P.A High-Temperature Chemical Kinetic Model of n-alkane Oxidation,JetSurF,Version 0.2.http://melchior.usc. edu/JetSurF/Version0_2/Index.html(accessed September 08, 2008).
(6)Muharam,Y.;Warnatz,J.Phys.Chem.Chem.Phys.2007,9, 4218.
(7)Wang,Q.D.;Wang,J.B.;Li,J.Q.;Tan,N.X.;Li,X.Y. Combust.Flame 2011,158,217.
(8) Li,J.;Shao,J.X.;Liu,C.X.;Rao,H.B.;Li,Z.R.;Li,X.Y. Acta Chim.Sin.2010,68,239.[李 军,邵菊香,刘存喜,饶含兵,李泽荣,李象远.化学学报,2010,68,239.]
(9)Tan,N.X.;Wang,J.B.;Hua,X.X.;Li,Z.R.;Li,X.Y.Chem.J. Chin.Univ.2011,32,1832.[谈宁馨,王静波,华晓筱,李泽荣,李象远.高等学校化学学报,2011,32,1832.]
(10)Wang,H.;You,X.Q.;Joshi,A.V.;Davis,S.G.;Laskin,A.; Egolfopoulos,F.N.;Law,C.K.USC Mech Version II. High-Temperature Combustion Reaction Model of H2/CO/C1-C4 Compounds.http://ignis.usc.edu/USC_Mech_II.htm(accessed May 2007).
(11) Holley,A.T.;You,X.Q.;Dames,E.;Wang,H.;Egolfopoulos, F.N.Proc.Combust.Inst.2009,32,1157.
(12)Sun,W.;Chen,Z.;Gou,X.;Ju,Y.Combust.Flame 2010,157, 1298.
(13) Kee,R.J.;Grear,J.F.;Smooke,M.D.;Miller,J.A.Chemkin-II: a Fortran Chemical Kinetics Package for the Analysis of Gas-Phase Chemical Kinetics.Report SAND89-8009.Sandia, 1989.
(14) Lutz,A.E.;Kee,R.J.;Miller,J.A.SENKIN:a Fortran Program for Predicting Homogeneous Gas Phase Chemical Kinetics with Sensitivity Analysis.Report SAND87-8248. Sandia,1987.
(15)Lu,T.F.;Law,C.K.Combust.Flame 2006,144,24.
(16) Jiang,Y.;Qiu,R.Acta Phys.-Chim.Sin.2009,25,1019. [蒋 勇,邱 榕.物理化学学报,2009,25,1019.]
(17)Wang,Q.D.;Fang,Y.M.;Wang,F.;Li,X.Y.Combust.Flame DOI:10.1016/j.combust ame.2011.05.019.
(18) Herbinet,O.;Marquaire,P.M.;Battin-Leclerc,F.;Fournet,R. J.Anal.Appl.Pyrolysis 2007,78,419.
(19) Vasu,S.S.;Davidson,D.F.;Hong,Z.;Vasudevan,V.;Hanson, R.K.Proc.Combust.Inst.2009,32,173.
(20) Shen,H.P.S.;Steinberg,J.;Vanderover,J.;Oehlschlaeger,M. A.Energy Fuels 2009,23,2482.
(21)Kee,R.J.;Grcar,J.F.;Smooke,M.D.;Miller,J.A.PREMIX:a Fortran Program for Modeling Steady Laminar One-Dimensional Premixed Flames.Report SAND85-8240.Sandia, 1985.
(22) Kee,R.J.;Warnatz,J.;Miller,J.A.A Fortran Computer Code Package for the Evaluation of Gas-Phase Viscosities, Conductivities,and Diffusion Coefficients.Report SAND83-8209.Sandia,1983.
(23)Ji,C.;Dames,E.;Wang,Y.L.;Wang,H.;Egolfopoulos,F.N. Combust.Flame 2010,157,277.
(24) Kumar,K.;Sung,C.J.Combust.Flame 2007,151,209.
(25)Kumar,K.;Mittal,G.;Sung,C.J.;Law,C.K.Combust.Flame 2008,153,343.
June 17,2011;Revised:September 27,2011;Published on Web:September 30,2011.
Mechanism Construction and Simulation for the High-Temperature Combustion of n-Dodecane
HUA Xiao-Xiao1WANG Jing-Bo1WANG Quan-De2TAN Ning-Xin1,*LI Xiang-Yuan1
(1College of Chemical Engineering,Sichuan University,Chengdu 610065,P.R.China;2College of Chemistry,Sichuan University,Chengdu 610064,P.R.China)
Using automatic generation software for hydrocarbon oxidation mechanisms,a detailed mechanism for the high-temperature combustion of n-dodecane was developed.A semi-detailed mechanism consisting of 202 species and 738 reactions was obtained by rate-of-production analysis while a skeletal mechanism including 53 species and 228 reactions was obtained using path flux analysis.Both the semi-detailed mechanism and the skeletal mechanism were validated by comparing simulation results including the ignition delay time,high-temperature pyrolysis,and the laminar flame speed with experiments.The current mechanism generation method and the generated semi-detailed and skeletal mechanisms for n-dodecane should be useful in computational fluid dynamics simulations.Finally,the major reaction pathways of n-dodecane oxidation and the important reactions in the ignition process were investigated by reaction path analysis and sensitivity analysis,respectively.
n-Dodecane;Combustion mechanism;Mechanism reduction; Chemical kinetic simulation
10.3866/PKU.WHXB20112755
∗Corresponding author.Email:tanningxin@scu.edu.cn;Tel:+86-28-85406139.
The project was supported by the National Natural Science Foundation of China(91016002).
国家自然科学基金(91016002)资助项目
O643
