6例强直性肌营养不良患者临床与病理分析
2011-08-11张文娟李秋香毕方方孙新刚尹小玲梁静慧
张 宁, 张文娟, 肖 波, 李秋香, 杨 欢, 毕方方, 孙新刚, 尹小玲, 梁静慧
强直性肌营养不良(Myotonic dystrophy,DM),是一种累及全身多系统的常染色体显性遗传疾病[1],男性多于女性,主要临床表现为肌强直、肌无力、肌萎缩,常伴有秃发、白内障、智力障碍、心律失常、糖尿病、周围神经受累等。本研究总结分析中南大学湘雅医院神经内科自2009年9月~2010年10月6例强直性肌营养不良患者资料,以期加强临床医生对该病的认识,进一步探讨其发病机制。
1 资料与方法
1.1 一般资料 6例患者均为男性,年龄8~43岁,平均26.2岁;发病年龄3~37岁,平均年龄14.5岁;病程4~40年,平均12年。仅有1例患者其爷爷奶奶为姑舅亲结婚。
1.2 临床表现 6例患者均慢性起病,有不同程度的肌紧张,肌强直,用筷、书写均不灵活,精细动作差,行走缓慢,静息时全身发紧,寒冷时加重,反复活动后缓解。其中1例以双手肌肉发紧、强直为首发症状,手握拳后松开困难,余5例均以双下肢肌强直首发,起步费力。4例患者叩击可见肌球征;仅1例有肌无力、肌萎缩,2例有脱发,1例伴智力障碍、吞咽困难、讲话不清,2例有张口费力。
1.3 实验室资料 6例患者血清肌酸激酶(CK)4例正常,2例轻度增高(CK 478.1 ~532.6U/L);肌电图均示肌源性损害,可见强直电位发放。
1.4 方法 6例患者均在局麻下行左肱二头肌开放式骨骼肌活体组织检查,取材标本经液氮-异戊烷快速冷冻,7μm冰冻连续切片,行苏木素-伊红(HE)染色、Gomori染色、PAS染色和油红(ORO)染色等特殊染色、还原型辅酶I四氮唑还原酶(NADHTR)染色、琥珀酸脱氢酶(SDH)染色、腺苷三磷酸环化酶(ATPase)、细胞色素C氧化酶(COX)、酸性磷酸酶染色、单磷酸腺苷脱氨酶(AMP)染色等酶组织化学染色,光镜下病理分析。
2 结果
光镜下病理改变:6例HE染色均可见:肌纤维大小不等,不同程度的肌纤维萎缩,1例可见角形纤维(见图1);5例可见不同程度的肌纤维变性坏死;6例未见明显再生肌纤维,1例可见肌分裂慢性病理改变;3例可见核内移、核聚集(见图2),1例有链状核(见图3);1例结缔组织重度增生,5例为轻度增生;间质均可见少量炎性细胞浸润;Gomori染色均未见破碎红纤维(RRF);PAS染色阴性;2例ORO染色可见部分脂肪代谢异常,余4例未见明显异常;AMP染色酶活性正常;NADH、SDH、COX可见酶活性局限性增高或减低,肌原纤维网紊乱,呈虫噬样改变(见图4);4例酸性磷酸酶染色酶活性增高;2例可见ATPase染色有肌纤维群组化,1例为I型肌纤维优势(见图5),另1例II型肌纤维群组化(见图6)。
3 讨论
DM是一种常染色体显性遗传异质性疾病,是成人最常见的一种肌营养不良,不仅仅有骨骼肌症状,还出现中枢神经系统症状(智力、情感障碍)、脱发、心脏传导速度异常、眼部症状(白内障、视网膜色素变性等)、内分泌异常(肾上腺、胰岛素分泌异常、甲状腺功能异常等)、周围神经受损,亦称萎缩性肌强直。
DM病因和发病机制的研究经历了长期的过程。目前认为其发病与离子通道有关[2]。正常肌细胞膜去极化引起钠离子通道短暂快速的开放,大量Na+内流,同时氯离子通道缓慢打开,Cl-进入细胞内使膜电位回到静息状态。钠离子通道突变引起Na+反复进入,重复引起去极化,如:高钾性周期性瘫痪、低钾性周期性瘫痪、波动性肌强直、恶性高热等。Cl-通道突变使Cl-进入减少,复极化减少,而去极化相对延长,肌肉反复放电从而引起肌强直[3,4],如:先天性肌强直、强直性肌营养不良等。Mankodi A[5,6]等学者先后在转基因动物模型和强直性肌营养不良患者中报道,CUG或CCUG重复扩增,其转录产物mRNA在肌组织中高表达,使跨膜Cl-通道水平减少,从而引起肌强直。
按基因突变位点和临床表现DM分为DM1和DM2两型,最近研究又发现 DM3型。1992年,Brook,Buxton等国外学者已确定DM1系第19号染色体长臂上(19q13.3)的DMPK基因突变,3’-端非翻译区的(CTG)n三核苷酸重复序列扩增,编码萎缩性肌强直蛋白激酶(dystrophia myotonic protein kinase,DMPK),然而这种酶的作用尚不明确,可能是在钙离子平衡稳态、信号转导以及骨骼肌纤维变性方面起作用。正常人该基因位点有4~37个CTG的重复序列,而DM患者的重复序列n可扩增至50~2000或更多至4000(见表1),其扩增程度与发病年龄及临床症状相关,重复单元越多,临床症状越严重。目前该蛋白酶致病动物模型已建立[7],最近国内赵晓萍[8]等人发现一个非CTG非CCTG重复扩展型强直性肌营养不良家系,该研究表明除CTG、CCTG短串联重复序列异常扩增外,可能存在新的DM致病基因。DM2型系 3号染色体长臂上(3q21.3)锌指蛋白9基因(ZNF9基因)的CCTG四核苷酸重复序列倍增突变引起[9],该基因编码锌指结构蛋白。正常情况下ZNF9基因位点有20~27个CCTG重复序列,DM2型患者可扩增达2000以上(见表1)。国外 Isabelle Le Ber[10]等发现一个新的致病基因突变位点,位于15号染色体长臂(15q21-24)上,该基因引起的肌病称为强直性肌营养不良DM3型。发病机制尚不十分明确。DM患者若母亲系基因携带者,后代具有发病提前和病情加重的遗传早现现象[11],因此,胎儿产前诊断意义突显重要。
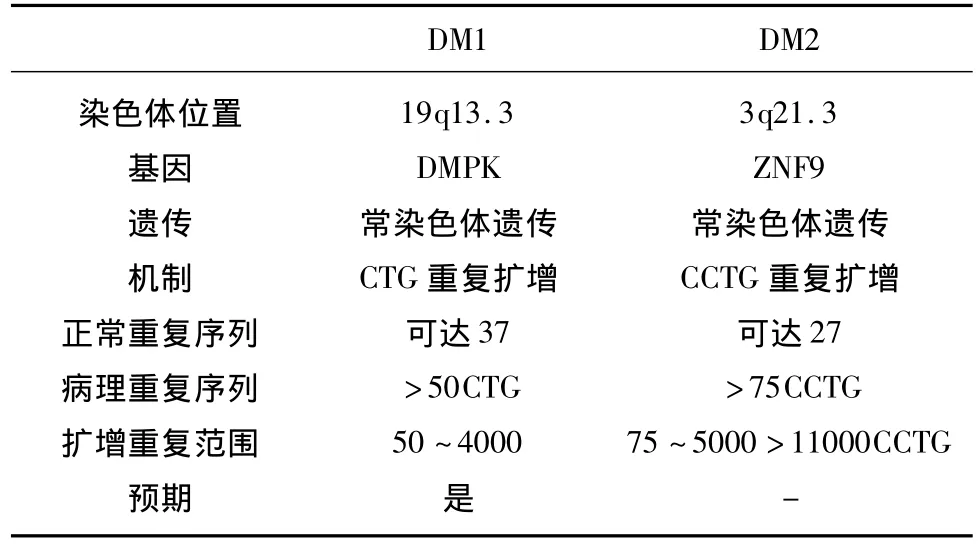
表1 DM1和DM2的病因
DM临床症状多种多样,DM1与DM2临床表现相似,共同的症状有肌强直、肌无力、肌萎缩、白内障、心脏传导改变、智力障碍、嗜睡、胰岛素抵抗、睾丸萎缩、肌痛、秃顶等[12]。本研究总结分析的6例患者临床表现除肌强直外,仅1例有肌无力、肌萎缩,余5例查体未见明显肌力低下及肌萎缩,患者主诉肌无力时可能为肌紧张、肌强直导致活动不灵活,精细动作不佳,并非真正无力。2例有脱发、秃头,无血糖升高、性激素等内分泌异常的表现。2例有张口费力,无吞咽困难、饮水呛咳,该患者张口费力可能为开始肌紧张,启动困难,反复张口活动后可缓解。1例其爷爷奶奶为姑舅亲结婚。无眼睛、心脏及消化道症状。DM1按发病年龄和疾病进展程度分3型:(1)经典型;(2)先天型;(3)轻型。经典型患者一般首发症状是远端肢体肌强直,本研究中有1例患者以双手肌肉发紧,强直为首发症状,符合这一经典临床特点。DM手部小肌肉萎缩有时出现较早,此时容易误诊,应注意与运动神经元病中的进行性脊肌萎缩症(SMA)、肌萎缩侧索硬化(ALS)、平山病、伴边缘空泡的远端型肌病等相鉴别(见表2)。部分DM患者可能会出现吞咽困难、构音障碍等[13],本研究中有1例伴吞咽困难、讲话含糊的患者;DM2早期表现近端肌受累,又称为近端型强直性肌营养不良,面肌较少累及,病变无特异性[12]。DM3型除有典型临床症状外,还伴有额颞叶痴呆[10]。本研究中1例有智力障碍的患者,其理解力、计算力等均差,此时应仔细询问病史,认真查体,结合其他临床特征及辅助检查以便做出诊断。DM晚期肌萎缩可累及近端,如累及面肌、眼外肌,导致面部表情减少,上睑下垂,颊部消瘦,导致“斧形脸”,是本病的特征性面容,本研究中6例患者均无此特征性表现。

表2 DM、青少年型SMA、ALS、平山病、伴有镶边空泡的远端型肌病比较
国内崔毅[14]等曾报道肌电图随病理特征不同,出现明显不同的电位,DM肌纤维病变早期,EMG表现自发电位、短棘波多相电位、复合性电位、神经传导速度(NCV)正常;中期,EMG可见肌强直电位、失神经复合型电位、群多相电位、短棘波多相电位和再生电位;晚期,EMG出现强直性电位、自发电位、复合性电位、失神经电位,呈神经性损害、肌源性损害,NCV均有减慢,部分未能引出神经动作电位,部分出现神经传导阻滞(包括MCV、SCV)。本研究中6例DM患者肌电图电生理检查均提示肌源性损害,有强直电位发放,可排除神经源性疾病,仅1例NCV减慢,余5例NCV在正常范围。血清肌酸激酶(CK)检测,4例患者CK正常,2例轻度增高(CK 478.1~532.6U/L)。
6例DM患者肌肉标本组织学及酶学病理变化,HE染色均可见肌纤维大小不等,不同程度的肌纤维萎缩,1例可见角形纤维(见图1),与神经源性病理改变的小角化肌纤维难鉴别,应紧密结合临床分析。6例均未见明显再生肌纤维,1例可见肌纤维分裂,肌分裂是代表肌原纤维的芽生,为慢性神经肌肉疾病的表现之一;3例可见典型核内移、核聚集(见图2),1例有链状核(见图3),均为DM病理的特征性变化,核聚集、核内移与链状核同时存在时,首先考虑DM这一诊断的可能性;2例ORO染色可见部分脂肪代谢异常,脂肪代谢包括脂肪酸的活化、转运和氧化等过程,其中多个环节异常均可造成脂肪酸代谢障碍,需紧密结合临床判断其是否为脂质沉积性肌病;NADH、SDH、CCO可见酶活性局限性增高或减低,肌原纤维网紊乱,呈虫蚀样改变(见图4),肌原纤维网紊乱、虫蚀样改变为细胞内酶的慢性改变;ATPase染色可见2例群组化肌纤维,1例可见选择性I型肌纤维萎缩及I型肌纤维占优势(见图5),另1例可见II型肌纤维群组化(见图6),余4例任意视野下均可见两型肌纤维呈镶嵌式分布。DM发病初期,I型与II型呈马赛克分布,随着疾病的进展,失神经支配,I型肌纤维逐渐占优势。DM病理变化的轻重与肌强直症状无明显相关性,与肌力低下的程度有关。DM的肌肉病理与临床表现相关,临床表现愈重肌肉损害愈明显[15]。分析本研究6例患者临床表现及病理结果,与该文献结果一致。
近年来由于分子生物学技术的发展,DM等遗传性肌肉疾病病理诊断已进入基因水平,基因检测为DM诊断的基本手段,但对经济条件要求很高,大部分医疗机构难普及,骨骼肌活检病理分析对此病的诊断有一定价值。
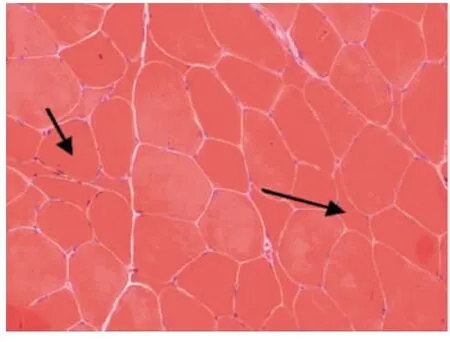
图1 肌纤维大小不等,箭头处为小角化肌纤维 HE×200
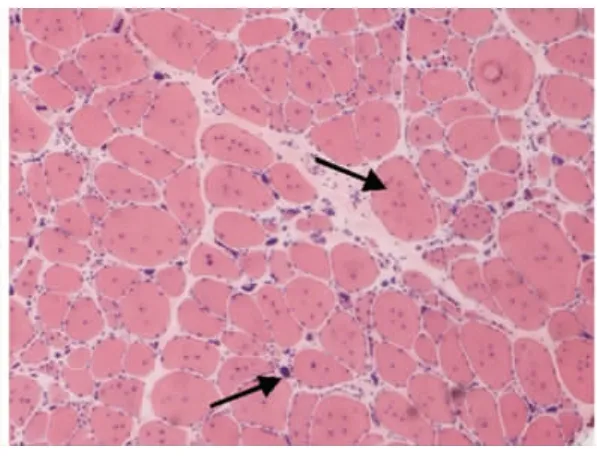
图2 箭头处为核内移,核聚集HE×100
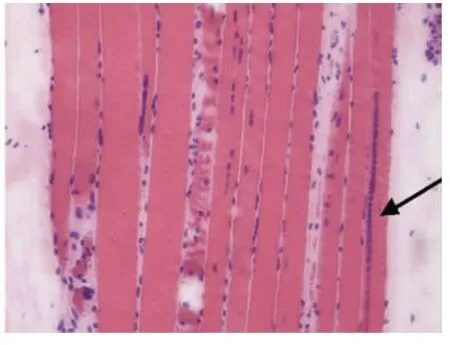
图3 箭头处为核聚集,链状核HE×200
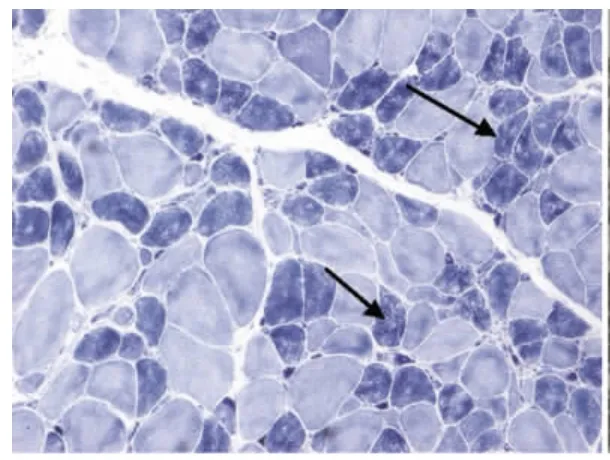
图4 肌原纤维网紊乱,虫噬状肌纤维NADH×100
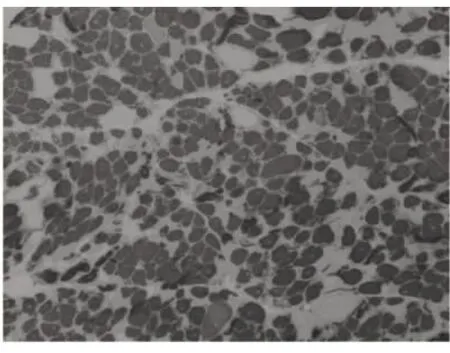
图5 Ⅰ型肌纤维群组化ATP4.3×100

图6 Ⅱ型肌纤维群组化ATP4.3×10
[1]Zerylnick C,Torroni A,Sherman SL,et al.Normal variation at the myotonic dystrophy locus in global human populations[J].Am J Hum Genet,1995,56(1):123 -130.
[2]Ranum LW,Day JW.Myotonic dystrophy:RNA pathogenesis comes into focus[J].Am J Hum Genet,2004,74(5):793 - 804.
[3]杨华艳,陈宜张.骨骼肌-神经系统离子通道病[J].基础医学与临床,2005,25(11):992 -998.
[4]江新梅,胡 静.离子通道与骨骼肌疾病[J].中国实用内科杂志,2007,27(6):876 -878.
[5]Mankodi A,Logigian E,Callahan L,et al.Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat[J].Science,2000,289(5485):1769-1773.
[6]Mankodi A,Takahashi MP,Jiang H,et al.Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscal in myotonic dystrophy[J].Mol Cell,2002,10(1):35 -44.
[7]Orengo JP,Chambon P,Metzger D,et al.Expanded CTG repeats within the DMPK 3’UTR causes severe skeletal muscle wasting in an inducible mouse model for myotonic dystrophy[J].Proc Natl Acad Sci USA,2008,105(7):2646 -2651.
[8]赵晓萍,谢惠君,郑惠民,等.一个非CTG非CCTG重复扩展型强直性肌营养不良家系[J].中华医学遗传学杂志,2004,21(5):459 -462.
[9]Christina LL,Kenneth R,Melinda LM,et al.Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9 [J].Science,2001,293(5531):864 -867.
[10]Le Ber I,Martinez M,Campion D,et al.A non-DM1,non-DM2 multisystem myotonic disorder with frontotemporal dementia:phenotype and suggestive mapping of the DM3 locus to chromosome 15q21-24[J].Brain,2004,127:1979 -1992.
[11]苏敬敬,王桂斌,赵武伟,等.CTG重复数与强直性肌营养不良的遗传早现现象[J].中风与神经疾病杂志,2003,20(4):337 -339.
[12]Schoser B,Timchenko L.Myotonic dystrophies 1 and 2:complex diseases with complex mechanisms[J].Curr Genom,2010(11):77 -90.
[13]De Swart BJM,Van Engelen BGM,Vande Kerkhof JPBM,et al.Myotonia and flaccid dysarthria inpatients with adult onset myotonic dystrophy[J].J Neurol Neurosurg Psychiatry,2004,75:1480 -1482.
[14]崔 毅,张永巍,王 晔,等.强直性肌营养不良的临床、肌肉病理与肌电图神经传导速度对比研究[J].临床神经电生理杂志,2002,11(2):79 -81.
[15]王 晔,郑惠民,谢惠君,等.强直性肌营养不良的临床与肌肉病理研究[J].中风与神经疾病杂志,2000,17(5):297 -298.
