老年痴呆药物研究进展
2011-05-17谢雨礼
谢雨礼
大冢(上海)药物研究开发有限公司,上海 201203
痴呆病(Dementia)是一种持续性的智能障碍,病因十分复杂,既有遗传因素,又有后天的环境影响和诱发,可引起痴呆的疾病高达百余种。其中,老年人常犯的痴呆主要有两种:一是老年性痴呆,即阿尔茨海默氏病(Alzheimer’s disease, AD);二是血管性痴呆(Vascular Dementia,VD)。这两种类型占到老年期痴呆的80%以上。老年痴呆病,由德国医生Alois Alzheimer于1906年首先发现,因此以其名字命名。这是一种慢性进行性的神经退行性疾病,早期表现为短期记忆力缺失,随着疾病的发展,大脑神经细胞功能逐渐丧失,造成记忆力、判断力、方位感、注意力和语言能力的损伤,并伴有行为和人格方面的改变,最终在5~10年内导致病人死亡。近年来,AD的发病率逐年攀升,已经成为严重威胁老年人生命健康和生活质量的主要疾病之一。根据阿尔茨海默氏病国际(AD International)的年度报告[1],2010年全世界年龄大于60岁的老人达7.5854亿,其中约4.7%、3556万人患有不同程度的AD。到2030年和 2050年,这个数字将大幅上升,分别达到6569万和11538万,其中尤以中国和印度等发展中人口大国增加最为迅速。在我国,大多数人认为中国的AD发病率低于西方国家,其实这是个误区,主要原因是人们一直认为记忆力衰退是人衰老的一部分,不是疾病,也很少到医院就诊。我国老年痴呆病的大规模调查表明,中国并非痴呆病的低发区,发病率与欧美发达国家相近,而且AD的发病率要明显高于血管性痴呆[2]。由于AD病人生活不能自理,长期需要专人照顾,因此给病人带来痛苦的同时,也给家庭和社会带来了沉重的经济和心理负担,已经成为不可忽视的社会问题。瑞典的Karolinska研究所2005年的一项研究估计,全球痴呆病的花费,如果包括非直接的费用,每年高达3000多亿美元,其中AD占到绝大部分[3]。AD非常顽固难治,其发病机制也不是很清晰,迄今尚未找到治愈该病的有效方法。目前AD一线治疗药物,比如乙酰胆碱酶(AChE)抑制剂,只能缓解早期病人的认知障碍,提供适度的症状改善作用,无法阻止病情的进展。因此寻找具有治疗作用 (disease-modifying)的抗AD药物成为当前药物开发的热点。由于AD带来的社会问题,这一领域也已引起世界各国政府的高度重视,成为优先支持的研究之一。近年来,神经生理、生化和药理等方面的基础研究不断深入,导致AD药物的研究和开发不断取得阶段性进展,但大多集中在临床前研究。最近几个寄予厚望的新药,如辉瑞的Latrepirdine和礼来的Semagacestat,三期临床相继失败,让人们重新开始审视AD药物的研发策略,主要集中在对AD发病机制的理解,诊断的缺陷,靶点的选择以及临床方案的设计等方面。本文将对AD的发病机制作简单介绍,并回顾基于这些机制的上市药物和临床研究药物,希望从这些成功和不成功的案例中得到启发,为今后AD药物的开发和研究提供思路和方向。
1 AD的发病机制
如图1所示,AD在病理方面具有两大显著特征:一是 β 淀粉样多肽(β-amyloid Peptide, Aβ)在大脑皮层和海马区神经细胞外累积沉淀形成β淀粉样斑(Aβ plagque);二是脑神经细胞内Tau蛋白异常聚集形成的神经原纤维缠结(neurofibrillary tangles)。另外,神经元突触功能异常,锥体神经细胞丢失以及乙酰胆碱等神经递质的大量降解也是较为常见的病理改变。

自20世纪90年代提出所谓的 “Amyloid Hypothesis”以来,人们普遍认为Aβ是AD发病机制的核心。具有神经毒性的Aβ在中枢神经系统累积诱导神经细胞凋亡,引起和促进一系列病理变化,最终导致AD复杂多样的病理特征和临床表现。因此以抑制和清除Aβ或其聚集的斑块为目的治疗手段成为当前药物开发的重要方向之一。尽管Aβ假说一直受到争议,但目前还没有其它理论能够替代它当前在AD研究领域的地位。首先,Aβ沉积是老年痴呆最典型的病理特征之一,Aβ沉积和疾病的发生和进展具有很强的相关性,通过老年斑诊断老年痴呆,准确率达到80%以上,另外10%~20%误诊一般都属于其它痴呆类型[4]。最近的研究表明,大脑区域的Aβ40和Aβ42含量几乎与AD病人临床上认知能力损伤具有定量的相关性,特别是在疾病的早期阶段表现尤为突出[5]。另外,已经成熟的Aβ蛋白可能不是唯一的致病因素,可溶性的、体积较小的Aβ分子寡聚体神经毒性更强,与神经细胞的损伤具有密切的相关性[6]。第二个强有力支持Aβ假说的证据是基因突变方面的。变异的Aβ前体蛋白(APP)或参与其切割的蛋白,如Presenilin 1和2的突变都会异常增加Aβ的产生,从而导致早期发生的老年痴呆或家族性老年痴呆(familiar AD)。虽然家族性老年痴呆在所有病例中只占到2%左右,但这些基因突变携带者几乎100%会患AD,强烈支持Aβ在AD发病机制中的关键作用[7]。大部分AD是所谓的零星发生的老年痴呆(Sporadic AD),一般发生较晚,遗传性也没有家族性AD这样强,然而有关基因的变异也能大大增加患病的可能性。目前研究最多的是一个与脂质代谢有关的基因ApoE,虽然ApoE变异致病没有象APP和Presenilin一样与Aβ直接相关,其他功能可能也参与作用,但ApoE变异确实也能加快Aβ的沉积[8]。转基因动物,特别是转基因小鼠过度表达APP等与Aβ相关的人类基因,能够重现AD的一些典型病理特征,而且表现出与年龄相关的学习和记忆力的衰退,成为研究AD和开发药物的有用工具,也为Aβ假说提供了大量的动物实验证据。然而,Aβ通过何种机制调节其细胞神经毒性还不是很清楚,有待进一步研究。
大部分基于Aβ假说的药物还处于各个阶段的研究当中,目前还没有成功上市的例子。倒是几个抑制Aβ产生的小分子药物和清除Aβ斑块的疫苗临床研究失败案例为Aβ假说的反对者提供了依据。其他与Aβ假说竞争的理论也因此日益受到重视。其中最具代表性就是以Tau为核心的致病机理。上述提到神经细胞里含有高度磷酸化Tau形成的神经原纤维缠结是老年痴呆另一个标志性病理特征。有研究表明,认知紊乱与纤维缠结的位置和含量有密切的相关性[9]。变异的Tau可导致前头侧头型痴呆(frontotemporal dementia),这表明 Tau 与痴呆症直接相关[10]。虽然这类痴呆在病理特征和临床症状上与AD有所不同,但表明Tau异常可以引起神经细胞丢失和认知紊乱,在AD中绝不只是病变结果这么简单。Tau是一种细胞质蛋白,与微管蛋白(Tubulin)有很高的亲和性,可以稳定神经元微管结构,保证神经元运输通道的畅通。在老年痴呆病人中,Tau被过度磷酸化,从而聚集成不溶的纤维缠结。这些纤维缠结有直接的神经毒性,也可消耗可溶性的Tau,阻断神经元的运输通道。两种因素都可能导致疾病的发生和恶化[11]。
线粒体功能紊乱 (Mitochondrial dysfunction)在AD中出现较早,常常导致神经细胞损伤和凋亡,被认为是神经性退变的原因之一[12]。APP和Aβ通过特殊运输机制,能够进入线粒体中与许多线粒体关键元件相互作用,破坏能量代谢和其正常功能。大脑炎症(Inflammation)是AD重要的临床表现之一。临床统计表明,服用非甾体抗炎药可减少患老年痴呆的风险[13]。最近一项基因筛选发现了CR1(complement receptor 1),它是一个调节自身免疫的关键基因,其突变是患老年痴呆的危险因素之一,从而建立了炎症和老年痴呆的基因链接[14]。其他AD的致病机制还包括活性氧化自由基(ROS)的大量产生对大脑分子的损伤;钙、铜、锌、钾等阳离子代谢紊乱;脂质和能量代谢紊乱等[15]。基于这些机制开发的药物,都已经进入临床研究。
2 AD的治疗药物
随着人口老龄化,老年痴呆的发病率迅速升高,其治疗药物市场前景巨大。据Research and Market的分析报告[16],2008年全球老年痴呆药物市场达到54亿美元,到2015年将会达到64亿美元。辉瑞、礼来等世界制药巨头都在加快抗AD药物的开发,以期在未来市场竞争中夺得先机。除上市药物外,截至2010年,世界药物研究开发信息库“Pharmaprojects”中收录处于临床的AD药物高达130余个,还有数百个处于临床前研究。这些药物包括小分子化合物、疫苗、抗体、特殊制剂,以及神经元再生和干细胞治疗等多种治疗手段。以下将对部分上市和处于临床研究的AD药物作一简要介绍。
2.1 上市药物
临床上用于治疗AD的药物大概有10余种,主要是拟胆碱药物和改善脑代谢的益智药。现已知脑内乙酰胆碱的含量与记忆力密切相关。老年痴呆病人大脑中乙酰胆碱大量减少,引起记忆力衰退,补充胆碱类物质能改善其记忆力和思维能力。直接给予胆碱或卵磷脂,由于代谢太快,不能增加乙酰胆碱的含量。而抑制乙酰胆碱水解酶可延缓乙酰胆碱的代谢分解,从而延长突触后受体的兴奋,改善记忆力。乙酰胆碱酶抑制剂是目前治疗AD的一线药物。
如图2所示,美国FDA批准的5个抗老年痴呆药物,其中4个为乙酰胆碱酶抑制剂,包括他克林(Tacrine)、多奈派齐(Donepezil)、利斯的明(卡巴拉汀,Rivastigmine)和加兰他敏(Galanthamine)。 国内研制的哈伯英或石杉碱甲,是我国学者从石杉属植物千层塔分离得到的一种生物碱,也是主要通过抑制乙酰胆碱酶达到改善记忆的疗效。乙酰胆碱酶抑制剂只能改善AD病人的症状,而不能延缓疾病的进程,主要对于早期AD病人疗效较好。盐酸美金刚胺是唯一用于中度和严重阶段AD的药物。美金刚胺的作用机制与乙酰胆碱酸抑制剂也有所不同,主要是通过阻断NMDA受体,延缓能损害神经细胞的兴奋神经递质谷氨酸(Glutamate)的释放。由于作用机制不同,美金刚胺可与乙酰胆碱酶抑制剂合用,增加疗效。比如,在一项由404位中度和严重的AD病人参加的三期临床研究中,美金刚胺和多奈派齐合用比单独使用多奈派齐取得明显更好的疗效[17]。这些药物除他克林由于肝脏毒性逐渐退出市场外,在国内都有销售。如图3所示,辉瑞的多奈哌齐目前在国内市场上处于领先地位,占到市场份额的58%左右。石杉碱甲和Forest Laboratories的美金刚胺市场份额相当,但美金刚胺应该更具前景。
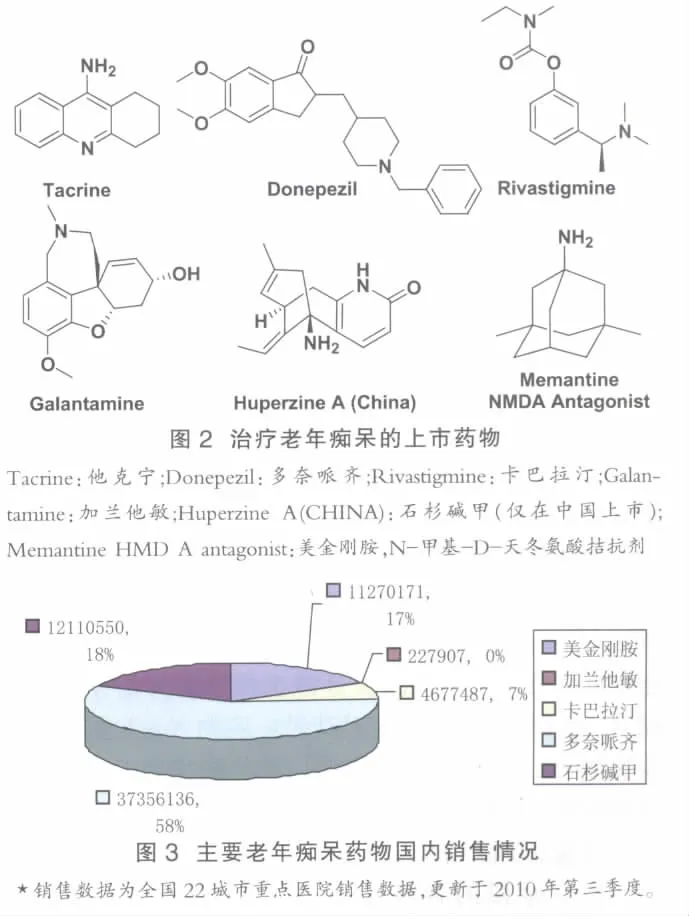
改善脑代谢的益智药一般具有扩张脑血管的作用,临床上常用于脑损伤后的恢复,这些药物对老年痴呆的某些症状如记忆力衰退、适应环境能力降低等有不同程度的改善作用。常用于老年痴呆的药物包括西坦类药物,吡硫醇,钙离子拮抗剂尼莫地平等。
2.2 处于临床研究的药物
目前上市的药物对老年痴呆都只能起到改善症状的作用。因此到目前为止,仍缺乏控制疾病进程的有效治疗手段。基于Aβ假说和Tau蛋白等新机制开发的药物都还处于临床研究阶段。
2.2.1 以Aβ为靶点的药物
2.1.1.1 降低Aβ产生的药物
APP代谢产生Aβ的过程是APP被β和γ分泌酶 (beta-and gamma-secretase)裂解为具有40、42或43个氨基酸的Aβ小肽。Aβ分子具有自发聚合的能力,它能在细胞外聚集成寡聚体、纤维和斑块等各种存在形式。因此抑制β和γ分泌酶成为阻止Aβ分子产生的有效手段(表1)。β分泌酶有许多底物,因此含有广泛的底物结合区域,另外抑制剂还需通过血脑屏障,开发其抑制剂很具有挑战性。目前只有5个化合物进入了一期临床。其中CoMentis开发的CTS-21166在健康成年人中口服给药,可以降低血浆中Aβ的含量,并且具有很好的耐受性[18]。有意思的是降血糖药物罗格列酮(Rosiglitazone)和吡格列酮 (Pioglitazone),通过刺激PPARγ受体可以抑制β分泌酶的表达,并促进APP的降解,从而减少Aβ的产生[19]。然而,临床研究表明,这些药物对AD病人的认知能力并没有改善作用[20],加之最近FDA对罗格列酮潜在心脏毒性的警告,PPARγ激动剂作为AD治疗药物的开发都已停止。
γ分泌酶负责Aβ产生的最后一步裂解,其抑制剂开发曾经是AD药物研发的热点之一。礼来的小分子γ分泌酶抑制剂Semagacestat(LY-450139)能够有效地减少Aβ的产生,进入到了三期临床研究。令人失望的是Semagacestat不但没有治疗作用,还加快病人认知功能的丧失,因此礼来于 2010年9月份宣布终止其临床研究[21]。另外几个药物,如Merck的MK-0752,辉瑞的 PF-3084014 和 GSI-9531(begacestat)也已终止临床研究。目前还有5个γ分泌酶抑制剂处于一期和二期临床研究。其中Mount Sinai医学院开发的NIC5-15是一个天然的单糖化合物,除增加胰岛素的灵敏性外,还能选择性的抑制γ分泌酶对Aβ的裂解作用,而对其另一底物Notch蛋白却没影响[22]。二期临床研究表明该药物治疗老年痴呆安全有效[23]。另外,部分非甾体抗炎药被发现可以调节γ分泌酶,抑制Aβ1-40和Aβ1-42的产生,而升高无害的Aβ1-38片段的含量。这些化合物中,Encore的Tarenflurbil(R-enantiomer of Flurbiprofen)进入到三期临床,但未能显示好的临床结果[24]。
有研究表明,升高alpha-分泌酶的活性,可以增加APP的良性代谢而降低Aβ的形成,而且其代谢产生的可溶性片段sAPP-alpha还有神经保护作用[25]。法国公司ExonHit开发的Etazolate是一种口服小分子药物,选择性调节GABA受体并可抑制PDE4,能够刺激alpha-分泌酶的活性,升高sAPP-alpha的产生。目前,该药物二期临床研究结束,但结果没有披露。美国Aphios开发的抗癌药物Bryostatin-1通过激活磷酸激酶PKC刺激alpha-分泌酶,促进sAPP-alpha的分泌,在老年痴呆动物模型中显示了较好的活性[26],该药物的二期临床研究正在计划当中。美国ProteoTech和我国的天士力公司共同开发的口服小分子抗老年痴呆药物Exebry-1在美国已经进入一期临床研究。该药物具有多种活性,除作用于Tau蛋白和抗炎外,也能调节beta和alpha分泌酶的活性,抑制Aβ的产生[27]。
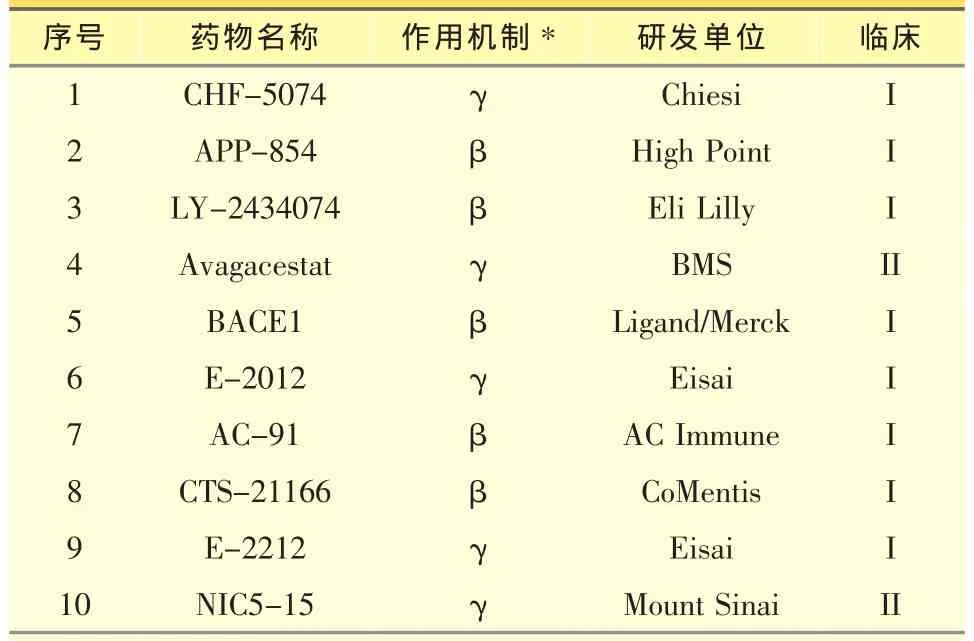
表1 处于临床研究的β和γ分泌酶抑制剂
2.2.1.2阻止Aβ聚合的药物
阻止Aβ分子聚合成具有神经毒性的寡聚体或斑块是基于Aβ假说开发AD药物的方向之一。抗聚合的化合物一般通过与Aβ单体分子结合,阻止其聚合和加快清除代谢。Bellus Health开发的tramiprosate是第一代的抗聚合药物。本品是口服有效的小分子化合物,倾向于与可溶性的Aβ分子结合而维持其非聚合状态,可惜该药物三期临床结果也不理想[28]。Bellus后于2008年将其开发成OTC营养品上市销售(商标名Vivimind)。 NRM-8499是该公司开发的tramiprosate前药,已进入一期临床。金属离子螯合剂可以与促进Aβ聚合的铜离子和锌离子结合,从而抑制Aβ聚合物的形成。Clioquinol(PBT1)是第一个进入临床研究的抗AD螯合剂,由于合成工艺问题,Prana Biotech放弃了该药物的开发,而将活性更好的类似物PBT2推上了二期临床。在一项为期12周的二期临床试验中,PBT2降低了大脑CSF中的Aβ1-42的含量,并改善了病人整体功能,本品的耐受性也非常好[29],不知什么原因该药物的三期临床研究一直没有启动。AZD-103是爱尔兰公司Elan开发的可透过血脑屏障的肌醇立体异构体,它可以与Aβ结合,调节Aβ分子的折叠,促进Aβ聚合物的分解。临床研究表明,本品高剂量(>1000mg·d-1)时有较大副作用,其低剂量(250mg·d-1)组的二期临床仍在进行中[30]。绿茶中得到的多酚化合物EGCg具有多种治疗AD的活性,其中之一就是抑制Aβ的聚合,其对早期AD病人的二期临床试验还在进行当中(NCT00951834)。
2.2.1.3 加速Aβ清除的药物
通过免疫的手段来清除Aβ是AD药物研发领域的重大突破之一。使用疫苗和抗体主被动免疫的策略,至少可通过三种机制清除Aβ:与Aβ结合,加速其聚合物的溶解;巨噬细胞的吞噬以及抗体的“提取”作用。Elan的科学家首次发现抗Aβ疫苗在动物实验中对AD有很好的治疗作用[31],随后该公司的Aβ疫苗AN-1792在二期临床研究中失败,主要是一部份病人发生了严重的脑部炎症。而且随后发现,虽然AN-1792降低Aβ的功能强大,甚至能完全清除病人脑中的Aβ老年斑,对病人认知功能却没有改善作用[32]。诺华的CAD-106,目前处于二期临床,能诱导特异的抗Aβ抗体但不引起CNS炎症,前景较为看好[33]。其他还有4个类似的疫苗处于早期临床阶段(表2)。值得一提的是,GSK和AFFiRis共同开发的AD-02是模仿天然Aβ的人工合成的小肽,能够诱导抗体特异性进攻Aβ的N端片断,在动物模型中疗效较好,目前已进入二期临床研究[34]。
被动免疫的策略是直接用抗体进攻Aβ分子,这种方法可以允许发展特异性更强的抗体,而且在老年病人中,这种方法可能更有效,因为老年人对疫苗的免疫反应能力普遍下降。目前至少有9个人源化的单克隆抗体处于临床研究,其中辉瑞的Bapineuzumab和礼来的Solanezumab已进入三期。Bapineuzumab能够与Aβ的N端片断紧密结合,主要进攻Aβ斑块,两项二期临床研究显示它能有效降低Aβ的含量,但对认知能力的改善没有统计学差异,似乎对不含 ApoEε4等位基因(ε4 allele)的个体更有效[35]。有意思的是,本品还可以降低CSF中Tau的含量[36]。该药物最大的副作用是血管源性脑水肿,几乎10%的临床参与者受到影响,而且副作用在携带有ApoEε4等位基因的个体中更为明显,不幸的是ApoEε4等位基因正是患老年痴呆的危险因素之一。目前,Bapineuzumab仍在进行三期临床研究[37]。礼来的Solanezumab主要作用于可溶性的Aβ分子,可加速Aβ的清除,而且能进攻几个Bapineuzumab无效的Aβ变种分子,本品能有效的降低脑中Aβ的含量[38],两项三期临床研究已经展开,将进一步研究其对早中期AD的疗效。另外,辉瑞的Ponezumab和罗氏的Gantenerumab也已进入二期临床研究,其它5个抗Aβ抗体还在一期进行安全性评价(表2)。

表2 处于临床研究的抗Aβ的疫苗和抗体
除生物制品外,也有少数小分子化合物被发现,可加速Aβ的清除。美国Archer公司开发的钙离子拮抗剂Nilvadipine和其类似物ARC-031就是例子之一,这些药物都还处在一期临床,疗效有待观察。
2.2.2 以Tau蛋白为靶点的药物Tau被认为在老年痴呆的致病机理中扮演重要的角色,因此也成为抗AD药物重要的靶点之一。目前主要有两种方法靶向Tau:一是抑制Tau的磷酸化,磷酸激酶调节的过度磷酸化是Tau异常聚合的主要原因;二是直接抑制Tau的聚合或促进其聚合物的分解。GSK-3是调节Tau磷酸化最主要的激酶。精神病治疗药物Valproate和锂被发现可抑制GSK-3,降低Tau的磷酸化程度,具有神经保护作用,可惜临床研究发现这两个药物对AD病人的认知能力和整体功能没有改善作用[39]。Noscira开发的非ATP竞争性GSK-3抑制剂Tideglusib目前仍在进行二期临床研究,还没有公开任何研究结果。亚甲基蓝是一种常用的染料,具有抗自由基的活性,并能直接抑制Tau的聚合,在动物实验中,单独或与乙酰胆碱酶抑制剂卡巴拉汀合用可扭转记忆力缺失和学习能力损伤[40]。临床研究表明,亚甲基蓝在60 mg剂量时可改善中期AD病人的认知功能,但100 mg组没有效果,被认为是剂型的缺陷造成的[41],TauRx公司最近又开发了其新的剂型TRx0037,已进入一期临床研究。Allon Therapeutics最近基于神经保护蛋白ADNP开发了一个八肽化合物Davunetide可调节微观蛋白,抑制Tau的磷酸化和聚合,二期临床试验表明,其对中等严重的AD病人有较好的疗效[42]。烟酰胺是维生素B3的生物活性体,研究显示该化合物可稳定微观结构,降低磷酸化Tau的含量,改善AD动物的记忆力[43]。
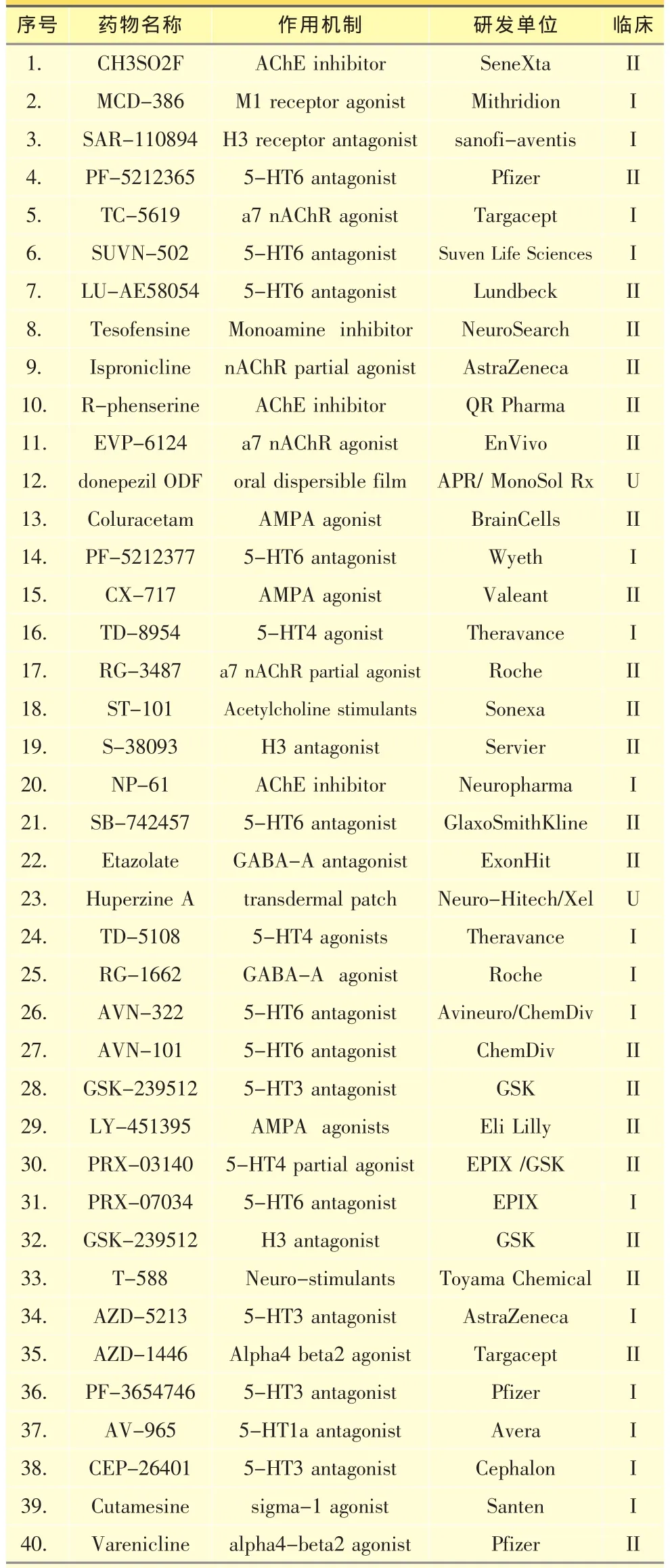
表3 处于临床研究的基于神经递质的药物
2.2.3 基于神经递质的药物神经递质紊乱与AD病理特征和记忆力衰退等临床表现直接相关。比如乙酰胆碱酶抑制剂就是基于AD中胆碱通路的严重缺失而发展的。这些药物依靠胆碱神经末梢起效,随着疾病的发展,这些末梢不断退化,因而此类药物也慢慢失去效果。神经递质乙酰胆碱参与的各种活动主要通过突触后膜两大受体系统发挥作用,即毒蕈碱型受体(M受体)和烟碱型受体(N受体)。发展这些受体的小分子激动剂成为研发AD药物的新方向。另外,5-羟色胺、组胺、AMPA、GABA和NMDA受体调节剂也日益受到重视。调节这些受体不但能改善认知能力,也可能降低Aβ和Tau起到治疗疾病的作用[44]。表3列出40个目前处于临床研究的基于神经递质的药物,其中包括石杉碱甲和多奈派奇等上市药物的新制剂,这些药物大多还处于一二期早期研究阶段,由于篇幅有限,本文不做详细介绍。
2.2.4 抗炎、抗氧化剂等其他作用机制的药物大量研究表明,抗炎和抗氧化剂对AD具有一定的治疗作用。非甾体抗炎药物,比如辉瑞的COX-2抑制剂Celecoxib用于老年痴呆的治疗,曾进入三期临床研究,但后来由于结果不理想被终止了。表4列出一些仍在进行临床研究的抗炎和抗氧化剂以及一些新作用机制药物 (如HDAC和PDE抑制剂等)。其中值得注意的是干细胞治疗 (HuCNS-SC,Stem-Cells)、细胞治疗(ECB-AD,NsGene A/S)和基因治疗(CERE-110,Ceregene)等新的治疗手段已经进入早期临床研究阶段,可能给AD的治疗带来新的希望。另外,几个具有神经保护等多种药理活性的植物提取物(INM-176,SK-PC-B70M,Anapsos)也在后期临床研究阶段,其治疗效果,令人拭目以待。

表4 处于临床研究的抗炎,抗氧化剂等其他作用机制的药物
结 语
随着基础研究日新月异,人们对AD的致病原因理解越来越深入。比如,Science最近报道一项研究结果表明,AD病人脑中Aβ分子的产生并没有受到影响,而是其清除受到抑制,并且发生远远早于疾病的临床表现[45]。因此,抑制Aβ分子产生的治疗方法需要重新考虑。这也表明,早期诊断对于及时治疗老年痴呆至关重要,等到疾病后期,大面积神经衰退可能难以逆转。另外,具有多种作用机制的药物,通过协同作用,可能比基于单个靶点或单种作用机制的药物更具前景。虽然,辉瑞的Dimebon(Latrepirdine)最近临床失败,但该药物通过多种机制,调节线粒体的功能而发挥治疗作用,这种靶向整个器官的新思路应该受到重视。特别是我国中药在这方面有一定的优势,如果能够合理设计临床方案,科学评价药物的疗效,中药在攻克AD的战役中可能会成为一支“奇兵”。
[1] Alzheimer’s disease international,World Alzheimer Report,2009,Executive Summary.
[2]Zhang ZX,Zahner GE,Roman GC,et al.Sociodemographic variation of dementia subtypes in China:Methodology and results of a prevalence study in Beijing,Chengdu,Shanghai,and Xian.Neuroepidemiology,2006,27(3):177.
[3] Wimo A,Winblad B,Jonsson L.An estimate of the totalworldwide societalcostsofdementia in 2005.Alzheimer’s and Dementia,2007,7:13.
[4] Newell KL,Hyman,BT,Growdon ET,et al.J.Neuropathol Exp Neurol,1999,58:1147.
[5] Lue LF,Kuo YM,Rocher AE,et al.Am J Pathol,1999,155:853.
[6] Lesne S,Koh MT,Kotilinek L,et al.Nature,2006,440:352.
[7]Hardy J,Orr H.J Neurochem,2006,97:1609.
[8] Cherbuin N,Leach LS,Christensen H,et al.Cognit Disord,2007,24:348.
[9] Thal D.Exp Neurol,2000,163:98.
[10] Hutton M,et al.Nature,1998,393:702.
[11]GoedertM,KlugA,CrowtherRJ.AlzheimersDis,2006,9:195.
[12]Reddy PH,Beal MF.Trends Mol Med,2008,14:45.
[13]McGeer PL,McGeer E.Neurobiol Aging,2007,28:639.
[14] Lambert JC,et al.Nature Genet,2009,41:1094.
[15] Citron M.Nature Reviews,2010,9:387.
[16] Medical news,http://www.news-medical.net/news/20091209/Key-trends-impacting-the- global-Alzheimers-dis ease-market.aspx.
[17]Forest Laboratories Inc,Medical release,http://www.frx.com.
[18]Tang JJN.Alzheiers Dement,2009,5:74.
[19] Landreth G,Jiang Q,Mandrekar S.Neurotherapeutics,2008,5:481.
[20] Gold M,Alderton C,Zvartau ME,et al.Alzheimers Dement,2009,5:86.
[21]Lilly press release,http://news-room.lilly.com,Sep 18,2010.
[22]WangJ,HoL,PassinettiGM.AlzheimersDement,2005,1:62.
[23] Grossman H,Marzloff G,Luo X,et al.Alzheimers Dement,2009,5:259.
[24] Green RC,Schneider LS,Amato DA,et al.JAMA,2009,302:2557.
[25]Van Marum RJ.Fundam Clin Pharmacol,2008,22:265.
[26] Etcheberrigaray R,Tan M,Dewachter I,et al.Pro Natl Acad Sci USA,2004,101:11141.
[27] Snow AD,Cummings J,Lake T,et al.Alzheimers Dement,2009,5:418.
[28] Aisen PS,Saumier D,Ferris S,et al.61st American A-cademy of Neurology annual meeting,Seattle,WA,USA.
[29] Lannfelt L,Blennow K,Zetterberg H,et al.Lancet Neurol,2008,7:779.
[30] Press release,Dec 15,2009,Elan.http://newsroom.elan.com.
[31]Schenk D,Barbour R,Dunn W,et al.Nature,1999,400:173.
[32] Holmes C,Boche D,Wilkinson D,et al.Lancet,2008,372:216.
[33] Winblad BG,Minthon L,Floesser A,et al.Alzheimer Dement,2009,5:113.
[34] Schneeberger A,Mandler M,Otawa O,et al.J Nutr Health Aging,2009,13:264.
[35] RinneJO,BrooksDJ,RossorMN,etal.Lancet Neurol,2010,9:363.
[36] Blennow K,Zetterberg H,Wei J,et al.Alzheimer Dement,2010,6:s134.
[37] Kerchner GA,Boxer Al.Expert Opin Biol Ther,2010,10(7):1121.
[38] SiemersER,Friedrich S,Dean RA,etal.Clin Neuropharmacol,2010,33(2):67.
[39] Tariot PN,Aisen PS.J Clin Psychiatry,2009,70:919.
[40] Deiana S,Harrington CR,Wischik CM,etal.Psychopharmacology,2009,202:53.
[41] Gura T.Nat Med,2008,14:894.
[42] Schmechel DI, Gerard G, Vatakis NG, et al.Alzheimers Dement,2008,4:T483.
[43] Green KN,Steffan JS.Martinez-Coria H,et al.J Neurosci,2008,28:11500.
[44] Fisher A.Neurotherapeutics,2008,5(3):433.
[45] Mauuenyega KG,Sigurdson W,Ovod V,etal.Science,2010,330:1774.
