中国汉赛巴尔通体分离株的多位点序列分型分析*
2011-01-24宋秀平栗冬梅黄儒婷李志芳刘起勇
赵 帆,宋秀平,栗冬梅,黄儒婷,李志芳,3,刘起勇
中国汉赛巴尔通体分离株的多位点序列分型分析*
赵 帆1,宋秀平1,栗冬梅1,黄儒婷2,李志芳1,3,刘起勇1
目的 了解中国汉赛巴尔通体的遗传背景和流行的常见亚型。方法对分离到的汉赛巴尔通体应用多位点序列分型方法(multilocus sequence typing,MLST),分析它们的分子流行病学特征和系统发育情况。结果80株汉赛巴尔通体一共分为3个序列型(sequence type,ST),其中ST1占90%(72/80),ST9占8.75%(7/80),另一个序列型为仅包含一株细菌的ST30;所有3个序列型均属于克隆群1(clonal comp lex 1)。结论与国外汉赛巴尔通体多位点序列分型结果相比,中国的序列型相对集中,说明中国汉赛巴尔通体的变异度和多样性较低,提示其进化相对缓慢。
汉赛巴尔通体;多位点序列分型;序列型;克隆群
汉赛巴尔通体(Bartonella henselae)是巴尔通体属(Bartonella)中一种重要的人兽共患病病原,为革兰氏染色阴性的需氧杆菌,其培养条件苛刻,分离难度很大。感染汉赛巴尔通体后,根据人体的免疫状况不同,身体会有不同的疾病反应:在免疫功能正常的患者中,汉赛巴尔通体主要引起猫抓病(cat scratch disease,CSD)[1-3],而免疫受损患者感染之后则可引起杆菌性血管瘤(bacillary angiomatosis)[4-5]、杆菌性紫癜(bacillary peliosis)[6-7]、培养阴性的心内膜炎[8]、视神经病变等严重疾病[9]。
由于病原菌的亚型往往与其致病的能力种类相关,在进行细菌致病的风险因素评估时考虑亚型与疾病的相关性,可以作出更切合实际的预防措施。而目前常用的16S rDNA分型方法仅能将汉赛巴尔通体分成2个亚型,并且两种都对人类致病[10-11],这促使研究者们选择更敏感的方法对汉赛巴尔通体进行分型。多位点序列分型(multilocus sequence typing,MLST)由 M aiden等研究设计,于 1998年首先应用于自然变异的脑膜炎奈色球菌(N eisseria meningitides)[12],后来被广泛应用于其他病原菌、环境菌和真核生物中。与传统分子生物学分型方法相比,MLST具有更高的分辨力,能将同种细菌分为更多的亚型,并确定之间的系统发育关系以及与疾病的联系[13],因而成为更有前景的检测和分型方法。2003年,MLST由 Iredell引入到汉赛巴尔通体的分型中[14],经A rvand优化后被广泛接受[15]。多年以来,各国研究者们使用MLST方法对汉赛巴尔通体进行了大量的分析,结果显示该方法能够较为合理地分析菌株间的遗传进化关系。为进一步了解中国的汉赛巴尔通体菌株的分子流行病学背景,我们对近几年分离到的汉赛巴尔通体进行了MLST基因分型,并分析了各序列型间的遗传关系及其与国外分离株间的联系。
1 材料与方法
菌株来源及培养 实验所用菌株为本科室自2005年至2010年间从采集到的家猫、家犬和人的全血中分离且经过鉴定的汉赛巴尔通体,共计80株。具体信息见表1。其中,78株猫源汉赛巴尔通体分离自北京市、河南省和山东省三个地区,一株犬源汉赛巴尔通体分离自北京,一株人源汉赛巴尔通体分离自山东。菌株的分离培养和复苏全部用含5%的无纤维新鲜羊全血琼脂糖培养基,在37℃、5%CO2的培养箱中培养。选择汉赛巴尔通体标准株 Houston-1[CIP 103737,G5436](A TCC 49882)为参考菌株。

表1 汉赛巴尔通体分离株基本信息Table 1 General information of B.henselae isolates in this study
1.2 MLST分析
1.2.1 基因组DNA的提取 复苏保存于-80℃超低温冰箱中的汉赛巴尔通体分离菌株,传代一次后收集纯培养的细菌菌体,使用 Qiagen公司的DNAeasy试剂盒提取全菌基因组DNA。
1.2.2 PCR扩增 根据文献中选用的8个管家基因,即 16S rDNA、batR、g ltA、f tsZ、gro EL、nlpD、ribC和rpoB,以及提供的引物序列合成8对扩增引物[14](表2)。引物的扩增条件使用标准菌株优化之后,应用于分离菌株的DNA模板,所有扩增产物经琼脂糖凝胶电泳检测为单一条带再进行DNA序列分析。引物合成由上海生工生物工程技术服务有限公司完成,测序工作由北京擎科新业生物技术有限公司完成。
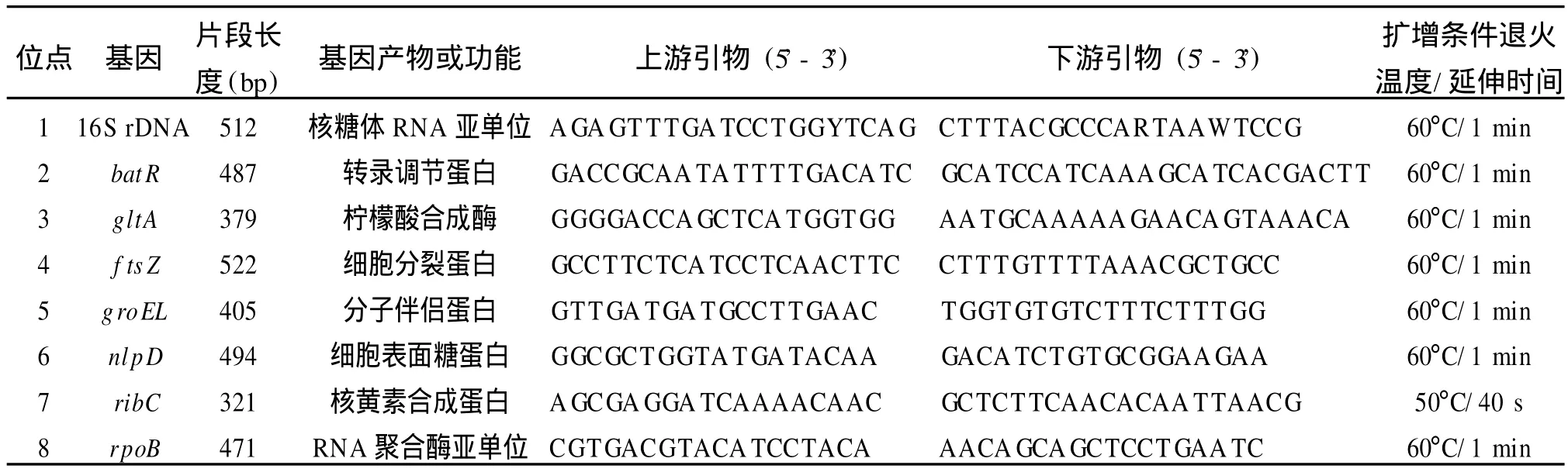
表2 MLST中所用8个管家基因及其扩增片段大小和引物情况Table 2 Primers used for am plification and sequencing of 8 housekeeping genes for B.hensela e MLST scheme
1.2.3 MLST结果分析 将8个管家基因片段的测序结果与NCB I中对应基因的所有等位基因型进行比较,分别确定每个分离株的等位基因编号,按照文献提供的顺序将各等位基因号依次排列获得每个分离株的等位基因谱,并确定各自的序列型。搜集国内外目前已经发表的所有序列型,应用eBURST程序分析中国汉赛巴尔通体分离株的序列型在克隆群(clonal comp lex)中的位置,应用M ega 4.0软件中邻接法 (neighbor-joining,NJ)基于核苷酸Kimura 2-parameter模型构建系统发育树。
1.3 统计分析 使用双侧 Fisher’s精确检验分析各序列型在分离地点间的差异,P<0.05被认为有显著性差异。
2 结 果
2.1 MLST结果
2.1.1 标准菌株的MLST基因片段电泳图谱 在优化的扩增条件下,用标准菌株DNA作模板进行PCR扩增,扩增产物通过琼脂糖凝胶电泳检测为单一条带,如图1所示。8对引物的扩增产物片段范围在300-500 bp之间,且经测序后比对完全与网上发布序列一致,为ST1,没有出现因为传代或扩增引起的突变。
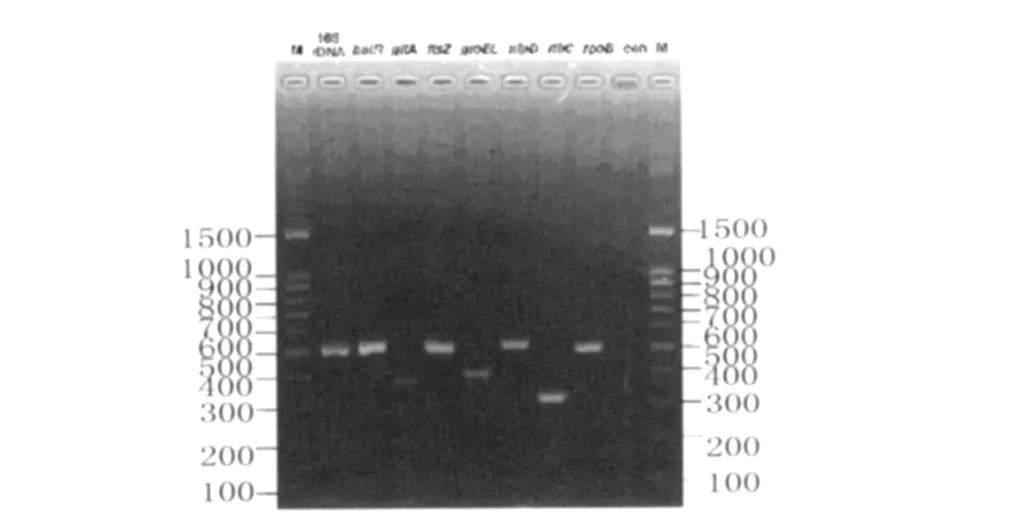
图1 标准菌株 Houston-1的8个管家基因片段扩增结果电泳Fig.1 Electrophoresis for amplified products of eight housekeeping genes from reference strain Houston-1 M:marker;con:negative control;PCR product for housekeeping fragments were show n as indicated
2.1.2 分离株的M LST结果 经过8对引物扩增和测序比对之后,80株分离株被分成3种序列型(ST1,ST9,ST30),其中72株为 ST1,占检测菌株的90%,在三个分离地点均有分布,包括唯一的犬源分离株和人源分离株;7株为ST9,占检测菌株的8.75%,全部为分离自北京的猫源汉赛巴尔通体;剩下的一株是新发现的序列型ST30,分离自山东省家猫,其管家基因 rpoB序列为首次报道的等位基因型6(allele 6),已经注册到美国国立卫生院的NCB I网站 (http://www.ncbi.nlm.nih.gov/)上,登录号为GU 477585.1,序列型ST30的等位基因谱信息也已注册到 MLST网站(www.m lst.net),可以在汉赛巴尔通体子数据库中找到。比较3种不同序列型菌株的地理分布(表3),发现ST1和ST9菌株的分离地点之间有显著性差异(P<0.05),而ST30因为其代表菌株数太少,所以其地理分布不具有统计学意义(P>0.05)。
2.2 MLST序列型的系统进化分析 根据MLST网站信息和文献报道,将已有的30种序列型及其等位基因谱(表4)录入eBRUST程序,分析各序列型间的系统发育关系,结果如图2所示,一共得到4个克隆群;A rvand M报道的独特性ST7[15]因为更多序列型的发现成为一个新的克隆群。分析中国目前分离株序列型间的相关性,发现序列型ST1、ST9和ST30聚集于克隆群1,其中,ST1和ST30亲缘性较近而与ST9相距较远。但是英国、德国、法国、意大利等欧洲国家分离得到了包括克隆群1~4在内的几乎所有序列型,与之相比,中国的汉赛巴尔通体序列型则单一而集中,种内多样性较低。

表3 MLST分析结果Table 3 MLST analysis result
8个管家基因的扩增片段按照相同顺序头尾相接串联形成一条新序列,将所有序列型各自串联得到的新序列用M ega 4.0软件的 neighbo r-joining(NJ)算法分析(图3)。所有序列型在NJ树状图中得到展现,从总体上显示出各序列型间的关系。其中,克隆群1所在分支经 bootstrap分析可信度为76%,说明聚集在该群中的序列型亲缘性相近,来源于同一个祖先的可能性较大;另外一个可信度为91%的分支,包含了克隆群4的所有序列型。而在eBURST结果中分属两个不同的克隆群(克隆群2和3)的序列型,在NJ树中却聚到一起,不过此分支的可信度不足50%,是否能被认可还需要更多数据的支持。

图2 eBURST分析各汉赛巴尔通体序列型间的系统发育关系Fig.2 Phylogenetic relationship between different B.henselae STsas determ ined by eBURST

表4 30个序列型信息Table 4 Profiles for 30 STs

图3 基于汉赛巴尔通体各序列型的串联序列构建的 NJ树(显示经bootstrap 1000分析可信度高于50%的节点)Fig.3 Neighbor-joining tree of the concatenated sequences of B.henselae STs(value of boostrap 1000 analysisgreater than 50%was shown)
3 讨 论
自2003年MLST应用于汉赛巴尔通体的分子分型研究以来,各国的相关研究工作陆续发表。Iredell从37株汉赛巴尔通体分离株中得到了7种序列型[14],几年之后,A rvand分析了收集自欧洲、美国和澳大利亚的182株汉赛巴尔通体,将序列型扩展到了14种[15]。2010年德国研究者新发现11种序列型[16],随后英国又发现了多个新的序列型而将总数增加到29。经过统计发现,序列型ST7仅局限于欧洲,ST1、ST5、ST6和 ST8呈世界性分布,但是其中ST5、ST6和ST8常见于欧洲,而 ST1正好相反,在欧洲的分离率较低。提示各序列型有一定的地理分布特点。
本文首次报道了中国汉赛巴尔通体分离株的MLST分析结果。从80株汉赛巴尔通体分离株中我们得到3种序列型(表3),其中世界性分布的序列型ST1占90%。说明汉赛巴尔通体ST1是中国的流行型,且呈广泛流行趋势。另外两种序列型分别占此次分析菌株的8.75%和1.25%,提示中国汉赛巴尔通体分离株序列型单一而集中、种内变异率低。结合 Yanagihara报道的数据[17],日本国内55株汉赛巴尔通体经MLST分析分为三种序列型,94.5%(52/55)的菌株为ST1,其余3株分属两个序列型,进一步提示汉赛巴尔通体在亚洲地区进化保守,群体中流行型别单一。
通过系统发育分析我们发现,中国汉赛巴尔通体的3种序列型均属于克隆群1,虽然ST9与ST1和ST30在克隆群中有一定距离(图2),但总体上该分支所包含的序列型还是属于亲缘性较高的一群汉赛巴尔通体,其bootstrap 1000分析可信度为76%,说明中国的汉赛巴尔通体虽然发生了少量变异,但是其变异仅在克隆群内部发生,主流序列型为ST1,菌株相对保守和集中,提示汉赛巴尔通体在中国的宿主群中可能尚未发生大规模的变异事件。
由于汉赛巴尔通体的研究在中国还属于初级阶段,可供参考的资料不多。并且临床医生对汉赛巴尔通体所致疾病不熟悉,临床报道仅限于部分医生的经验而少有实验室证据。本文利用近年来从宿主(主要为家猫)中分离到的汉赛巴尔通体菌株,分析其多位点序列分型结果,发现中国家猫中汉赛巴尔通体的流行型ST1,是已被广泛报道的致病亚型,并且在家犬和人血中分离到的菌株也属相同的序列型,提示中国存在汉赛巴尔通体感染的危险因素,其感染状况可能被严重忽视,人群中的汉赛巴尔通体流行型有待进一步研究核实。
[1]Jones PD.Cat scratch disease and Rochalimaea henselae[J].Med J Aust,1993,159(3):211.
[2]Margileth AM,Hayden GF.Cat scratch disease.From feline affection to human infection[J].N Engl JMed,1993,329(1):53-54.
[3]Margileth AM.Cat scratch disease[J].Adv Pediatr Infect Dis,1993,8:1-21.
[4]Koehler JE,Quinn FD,Berger TG,et al.Isolation of Rochalimaea species from cutaneous and osseous lesions of bacillary angiomatosis[J].N Engl J Med,1992,327(23):1625-1631.
[5]Koehler JE,Cederberg L.Intra-abdominalmass associated with gastrointestinal hemorrhage:a new manifestation of bacillary angiomatosis[J].Gastroenterology,1995,109(6):2011-2014.
[6]Tappero JW,Mohle-Boetani J,Koehler JE,et al.The epidemiology of bacillary angiomatosisand bacillary peliosis[J].JAMA,1993,269(6):770-775.
[7]Koehler JE.Bartonella-associated infections in H IV-infected patients[J].A IDSClin Care,1995,7(12):97-102.
[8]Bookman I,Scholey JW,Jassal SV,et al.Necrotizing glomerulonephritis caused by Bartonella henselae endocarditis[J].Am J Kidney Dis,2004,43(2):25-30.
[9]Depeyre C,Mancel E,Besson-Leaud L,et al.Abrupt visual loss in children.Three case studies of ocular bartonellosis[J].J Fr Ophtalmol,2005,28(9):968-975.
[10]Bergmans AM,Schellekens JF,van Embden JD,et al.Predominance of two Bartonella henselae variants among catscratch disease patients in the Netherlands[J].J Clin Microbiol,1996,34(2):254-260.
[11]Mainardi JL,Figliolini C,Goldstein FW,et al.Cat scratch disease due to Bartonella henselae serotype Marseille(Swiss cat)in a seronegative patient[J].J Clin Microbiol,1998,36(9):2800.
[12]Maiden MC,Bygraves JA,Feil E,et al.Multilocus sequence typing:a portable app roach to the identification of cloneswithin populations of pathogenic microorganisms[J].Proc Natl Acad Sci U S A,1998,95(6):3140-3145.
[13]Maiden MC Multilocus sequence typing of bacteria[J].Annu Rev Microbiol,2006,60:561-588.
[14]Iredell J,Blanckenberg D,A rvand M,et al.Characterization of the natural population of Bartonella henselae by multilocus sequence typing[J].J Clin Microbiol,2003,41(11):5071-5079.
[15]A rvand M,Feil EJ,Giladi M,et al.Multi-locus sequence typing of Bartonella henselae isolates from three continents reveals hypervirulent and feline-associated clones[J].PLoS One,2007,2(12):1346.
[16]Mietze A,Morick D,Kohler H,et al.Combined MLST and AFLP typing of Bartonella henselae isolated from cats reveals new sequence types and suggests clonal evolution[J].Vet Microbiol,2011,148(2-4):238-245.
[17]Yanagihara M,Tsuneoka H,Hoshide S,et al.Molecular typing of Bartonella henselae DNA extracted from human clinical specimens and cat isolates in Japan[J].FEMS Immunol Med Microbiol,2010,60(1):44-48.
Multilocus sequence typing analysis for Bartonella henselae isolates in China
ZHAO Fan,SONG Xiu-ping,L IDong-mei,HUANG Ru-ting,L IZhi-fang,L IU Qi-yong
(State Key Laboratory for Infectious Disease Prevention and Control,National Institute for Communicable Disease Control and Prevention,Chinese Center for Disease Control and Prevention,Beijing 102206,China)
To understand the genetic background and epidemiological subtype of Bartonella henselae isolates in China,multilocus sequence typing method was used to analyzed the molecular epidemiological characteristic and phylogenetic relationship for B.henselae.Results showed that 80 B.henselae isolates were belonged to three sequence types(STs).90%isolates was ST1(72/80)and 8.75%was ST9(7/80),whileonly one strain was belonged to a new type ST-ST30.In addition,all the three STs were belonged to clonal complex 1.Compared with the other reports on B.henselae MLST analysis,the STs were centralized in China,suggesting low diversity and evolution speed among B.henselae isolates in China.
Bartonella henselae;multilocus sequence typing;sequence type;clonal comp lex
R181.2
A
1002-2694(2011)07-0592-05
*重要病媒生物监测和传播相关病原体检测技术研究(2008ZX10004-010);传染病防治科技重大专项“传染病监测技术平台”细菌性传染病病原体流行规律及变异研究(2009ZX10004-203)
刘起勇,Email:liuqiyong@icdc.cn
1.中国疾病预防控制中心传染病预防控制所媒介生物控制室,传染病预防控制国家重点实验室,北京 102206;2.北京市丰台区疾病预防控制中心,北京 100071;3.郑州大学公共卫生学院流行病与卫生统计学教研室,郑州 450001
2011-01-16;
2011-04-24
