基于Maize6H-60K芯片精准分型的玉米DH群体遗传规律研究
2022-11-07马世鹏张云龙段民孝易红梅许理文葛建镕范亚明赵怡锟田红丽霍永学王凤格
马世鹏 张云龙 段民孝 易红梅 许理文 葛建镕 范亚明 赵怡锟 田红丽 杨 扬 霍永学 王凤格 王 蕊* 李 莉
(1.中国农业大学 农学院,北京 100193; 2.北京市农林科学院 玉米研究所,北京 100097)
玉米(Zea
mays
L.)作为全球重要的作物及研究模式植物,在常规育种过程中纯系的获得至少需要2~4年,耗费大量人力与资源。DH群体作为永久遗传群体,可以在不同的环境或年限进行重复试验,保证结果的准确性,被广泛应用到玉米、小麦、水稻等作物QTL定位研究中。染色体重组交换既是进化的根本动力也是形成遗传多样性的根本原因。群体中个体出现的有效重组交换可以提高QTL精确定位的效力,同时也为育种工作提供了遗传基础。已有研究证实染色体上交换重组热区由甲基化、雌雄配子体等因素共同决定。而由于低重组交换区域缺乏重组交换,有害突变会在此类区域世代积累,导致使用传统育种手段消除有害突变非常困难。因此从遗传作图和建立育种群体2个角度都需要明确DH群体的重组交换规律。偏分离现象是指位点基因型频率不符合孟德尔分离比例的情况,逐渐被公认为是物种进化的重要动力之一。在使用分子标记绘制不同遗传群体遗传图谱过程中,普遍会发现偏分离现象。Lu等研究将多数偏分离标记聚集的染色体区域称为分离畸变区域(Segregation distortion regions,SDRs)。随着高密度SNP芯片在遗传群体中的应用,为全基因组水平呈现遗传群体的偏分离现象提供了机会。SNP标记作为第三代分子标记,不仅具有数量多、分布广、多平台数据易整合等优势,而且可以利用基因芯片和测序技术完成高密度位点的基因分型,使结果更加准确。其中芯片比起基于测序基因分型(genotyping-by-sequencing,GBS),具有成本低、流程成熟简单、数据运算量小等优点;与低深度测序相比,芯片使用固定的检测位点,注释信息详细,参考资料丰富。北京市农林科学院玉米研究所报道了Maize6H-60K SNP芯片研制过程及其在玉米种质资源评估、分子鉴定、分子育种中的应用。数据分型是基因芯片技术的一个重要环节, 其可靠性与准确性直接影响芯片的推广应用,基因分型技术的必要手段便是利用分子标记实现对基因型数据的准确采集,低质量的基因型数据会影响统计效力和偏差判断,但大多数遗传研究默认采集的基因型数据是准确的,例如在QTL定位研究中,基因分型错误导致稀有等位基因的分析结果稳定性较低;在绘制SNP高密度连锁图谱过程中,基因分型错误还会导致计算连锁不平衡和标记遗传距离出现偏差。虽然芯片技术发展逐渐成熟,已形成高通量、自动化、标准化的流程,然而基因分型错误仍然是目前高密度芯片应用过程中主要问题之一。目前,缺乏精准采集基因分型数据的研究,削弱了研究结果的可信度。而DH群体具有背景高度纯合的特点,预期只包括2 种纯合基因型,分型结果的可预测性强,在研究基因分型问题上发挥着独特的作用。
目前,大部分研究仅报道个别的重组交换热点及分离畸变区域,利用精准分型的高密度SNP芯片解析DH群体遗传规律的研究鲜见报道。本研究针对DH群体背景高度纯合的特点,利用Maize6H-60K高密度SNP芯片及自主研发的精准分型策略得到DH群体的精准分型数据,旨在精准解析DH群体重组交换及分离规律,以期为玉米DH群体在育种中的应用提供参考。
1 材料与方法
1.1 材料
试验材料包括209份‘先玉335’DH群体及‘先玉335’双亲‘PH6WC’和‘PH4CV’,由北京市农林科学院玉米研究所提供,用于分析该群体的遗传规律。另设置17份在不同芯片板上的试验重复,用于验证精准分型结果的重复性和准确性。
1.2 试验方法
1
.2
.1
样品准备和芯片试验使用改良CTAB法提取基因组DNA并去除RNA,用1%琼脂糖凝胶电泳和紫外分光光度计(Nanodrop 8000,美国Thermo Fisher公司)检测DNA质量和浓度,确保DNA主带清晰无降解,并统一稀释到50 ng/μL备用。
利用Maize6H-60K芯片对供试样品进行高密度基因分型(GeneTitan平台,Affymetrix)。试验流程参照Affymetrix公司提供的操作指南(https:∥www.thermofisher.cn获得),完成DNA扩增、片段化及沉淀、干燥、悬浮、质量控制及变性、杂交、清洗、染色、扫描等流程。
1
.2
.2
初始基因分型使用Axiom Analysis Suite软件(Thermo Fisher Scientific Affymetrix,V4.0.3.3,以下简称AxAS软件)分析原始信号CEL文件并获得基于软件的基因分型结果。使用Best practices workflow流程分析,其中自交系分析参数选择16,对所有样品进行分型,其他阈值参数为默认参数Diploid.v4(https:∥www.thermofisher.cn获得)。
1
.2
.3
精准分型策略使用本实验室研发的精准分型策略对筛选后的所有位点重新分型。精准分型策略主要步骤为:
1)使用高斯混合模型进行聚类。首先对原始荧光信号S
进行标准化处理:使用S
、S
定义原始荧光信号A、B的值,并且分别定义二维坐标系X
、Y
。用N
(N
,N
)定义标准化计算后的S
、S
,其中N
、N
的转换公式如下:N
=logS
-logS
(1)
N
=(logS
+logS
)/
2(2)
其次对位点数据进行高斯混合模型,使用贝叶斯信息准则以及“肘部原则”进行评估,确定最优聚类数并导出分型结果。
2)依据历史数据确定基因分型。根据骨干自交系聚类结果统计了芯片所有位点纯合聚类的中心点坐标和标准差范围。参照自交系中心点坐标预测DH群体纯合聚类中心点坐标,剩余的数据点根据Y
轴相对坐标区分杂合和缺失:Y
Y
)(3)
Y
>Average(Y
+Y
)(4)
式中:Y
为缺失数据中心点的N
;Y
为杂合数据中心点的N
;Y
为AA纯合数据中心点的N
;Y
为BB纯合数据中心点的N
。3)分型结果验证。根据同一聚类的样品横纵坐标值分别计算平均值和方差,筛选差异较大的个别位点进行人工审核。
1.3 统计分析
基于精准分型结果使用SNP比对统计工具软件(V1.0,软件登记号:2018SR026743)进行重复样品比对和不同分型方法结果比对,统计差异位点个数。以1 Mb大小为窗口计算区间内的标记个数,使用Rstudio(V1.4.1106)中Complex Heatmap包绘制标记密度热图。使用Python统计‘先玉335’DH群体全基因组水平的重组交换点。使用Origin 2021绘制10条染色体的标记分离系数散点图,其中标记分离系数=与母本基因型相同个体数/群体个数,将标记实际观察值按孟德尔理想分离比(1∶1)进行χ
检验,判断标记发生分离的比例和方向。2 结果与分析
2.1 适用于DH群体遗传研究的标记筛选
适用于DH群体遗传研究的标记特征为双亲具有不同的等位基因,且基因芯片分型为2个清晰的聚类,分别为AA和BB纯合子(图1(a))。剔除不涉及分型问题和遗传规律的3种类型位点:1)由于样品混杂、剩余变异等原因,出现杂合聚类的标记(图1(b));2)单态型位点(图1(c));3)样品缺失率>5%的标记(图1(d))。最终从61 214个SNPs中保留19 620个SNPs,其中由于质量差而被剔除的标记仅占总标记的1.93%,说明Maize6H-60K芯片原始数据质量高,能够用于后续研究。
2.2 精准分型策略及标记分布
将筛选出的SNP标记分型误判原因主要归纳为3种类型:1)将杂合聚类误判为纯合聚类(图2(a));2)缺失数据误判为纯合聚类(图3(a));3)杂合和缺失的聚类被误判为2种纯合聚类(图4(a))。
使用精准分型策略对19 620个SNPs重新聚类,通过比较AxAS软件与精准分型结果显示2种不同分型错误的SNP标记均能正确分型(图2~图4)。出现在杂合基因型位置的数据点被正确判断为杂合聚类(图2(b)),且当出现缺失数据和杂合基因型数据时,不会和纯合基因型聚类混淆(图3(b)和图4(b))。通过比对不同芯片板上的试验重复评估精准分型结果的一致性,结果显示重复样品间平均差异标记数为1.8 个,一致性达到99.9%,其中5对结果一致性达到100%。以上说明精准分型数据具有更高的准确性和稳定性。
进一步剔除精准分型校正后出现缺失和杂合数据的标记,最终得到入选标记为18 947个SNPs,约占芯片全部位点的31%。入选标记在全基因组上的分布与染色体长度呈正相关,在1号染色体分布的个数最多为2 940,10号染色体最少为1 357个,标记间的平均距离为0.097~0.118 Mb(表1),除染色体着丝粒附近区域外均匀覆盖染色体(图5),说明入选标记能够有效跟踪基因组所有区段的重组交换。
2.3 ‘先玉335’DH群体的重组交换规律
由图6和图7可知,只有1号染色体发生2次交换的个体比例(33%)高于发生1次交换的个体比例(29%)(图6);在2~9号染色体上发生1次交换的个体比例最高(46%~35%);有12%~34%的个体在单条染色体上不发生交换,其中6号染色体最高(34%),其次为10号染色体(29%)。10条染色体的平均交换次数与染色体长度呈正相关,从1号染色体的1.84次到10号染色体的1.03次不等(图7),其中6号染色体平均交换次数小幅度降低。
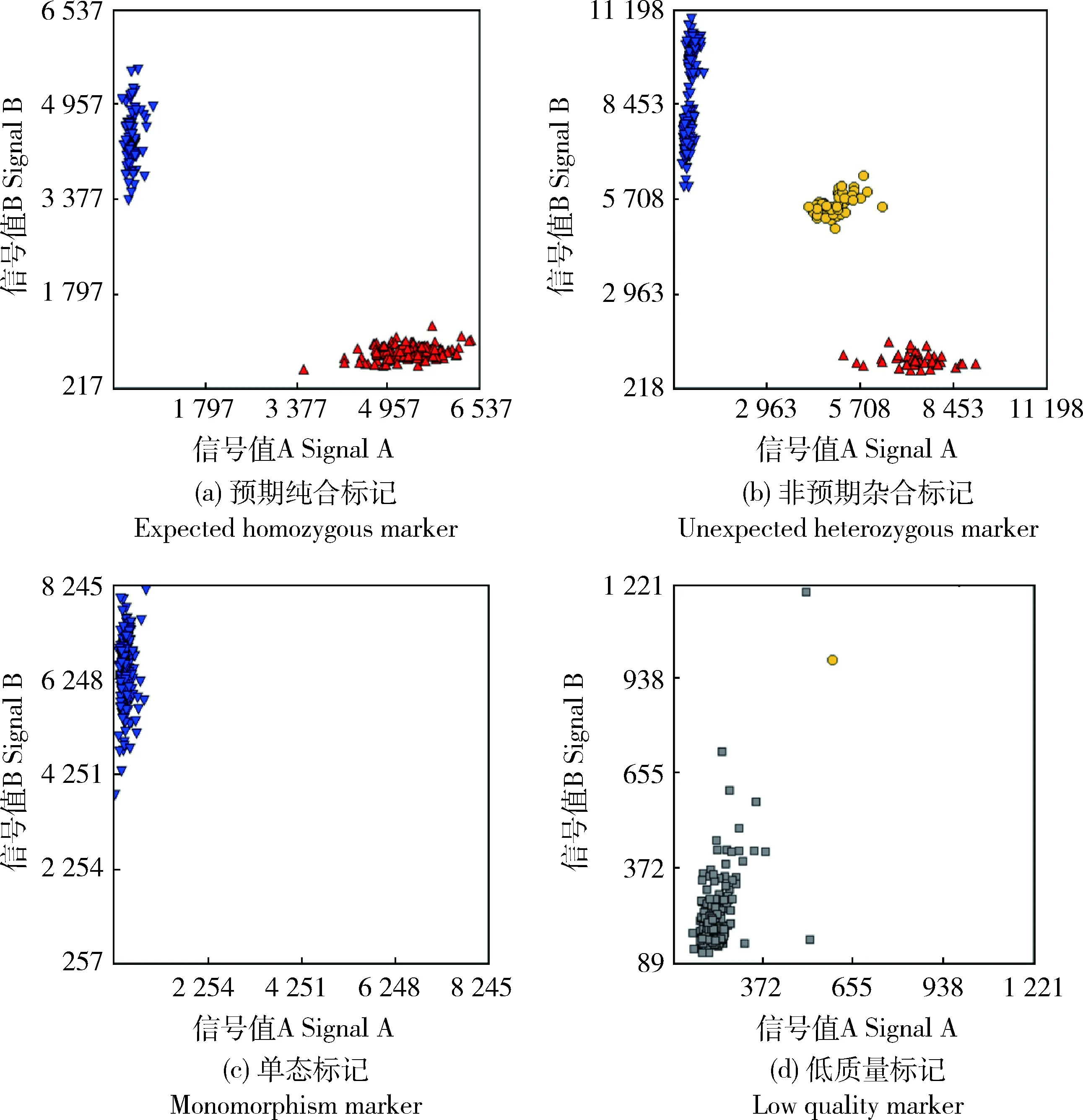
红色三角形表示AA基因型,蓝色三角形表示BB基因型,黄色圆形表示AB基因型,灰色方块表示缺失数据点。下同。 Red triangular represents AA genotype, blue triangular represents BB genotype, yellow circle represents AB genotype, gray square represents the missing data points. The same below.图1 SNP基因芯片的标记表现类型Fig.1 Performance types of markers on SNP gene array
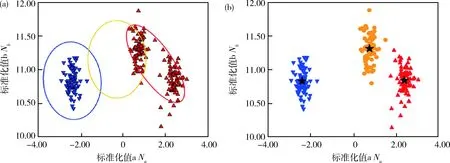
黑色星形标识代表聚类中心点。下同。 Black star logo represents the cluster center. The same below.图2 AX-91094685的AxAS软件(a)和精准(b)分型对比Fig.2 Comparison of AxAS software (a) and precise (b) typing of AX-91094685
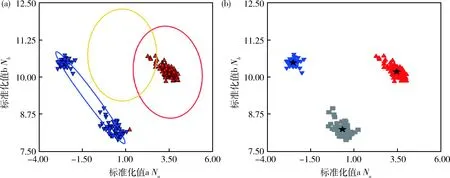
图3 AX-108052387的AxAS软件(a)和精准(b)分型对比Fig.3 Comparison of AxAS software (a) and precise (b) typing of AX-108052387
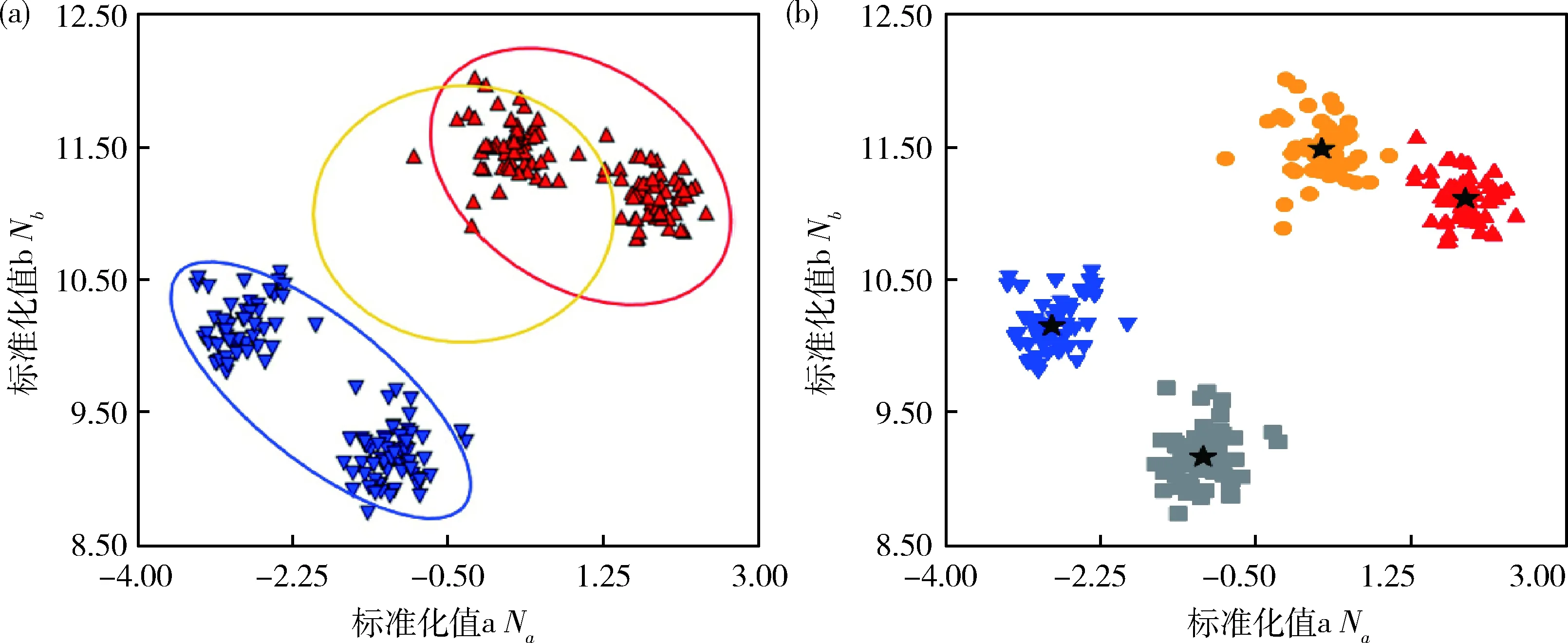
图4 AX-108103101的AxAS软件(a)和精准(b)分型对比Fig.4 Comparison of AxAS software (a) and precise (b) typing of AX-108103101
表1 SNP标记在基因组上的分布
Table1 Distribution of SNP markers in the genome
染色体Chromosome标记数量Number ofmarkers起始物理位置(Mb)Start physicalposition终止物理位置(Mb)End physicalposition区间大小/MbInterval size平均距离/MbAveragedistanceChr 12 940.0000.086299.773299.6870.101Chr 22 245.0000.133237.791237.6580.106Chr 32 159.0000.304232.221231.9170.104Chr 42 047.0002.838241.304238.4660.116Chr 52 126.0000.098217.799217.7010.097Chr 61 563.0000.363168.385168.0220.107Chr 71 574.0000.036176.810176.7740.112Chr 81 474.0000.021174.923174.9020.118Chr 91 519.0000.314156.873156.5590.101Chr 101 357.0001.284149.576148.2920.109
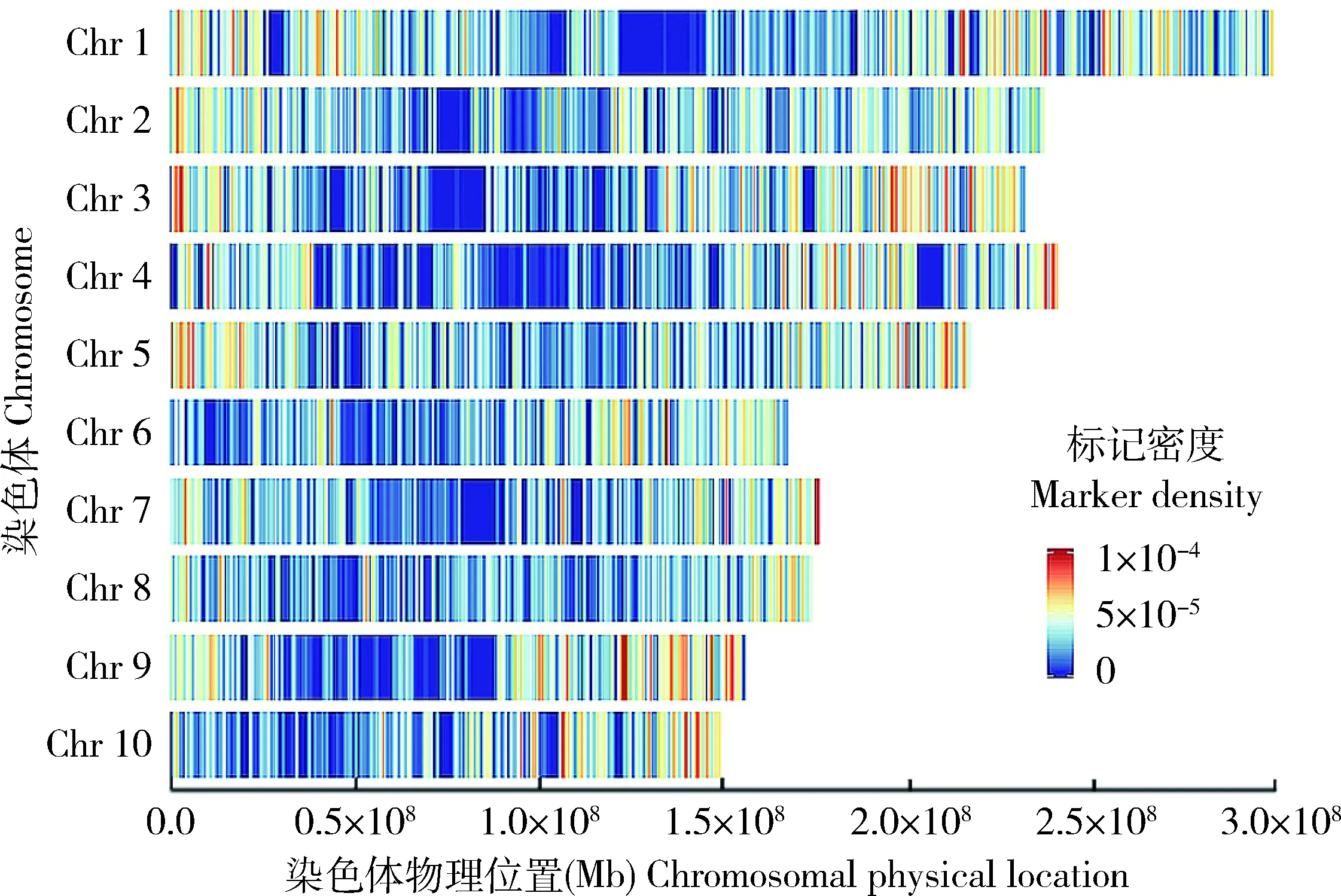
以1 Mb为窗口大小,计算每个窗口的标记密度。 Take 1 Mb as the window size, the marker density was calculated for each window.图5 入选标记分布密度Fig.5 Distribution density of markers on the genome
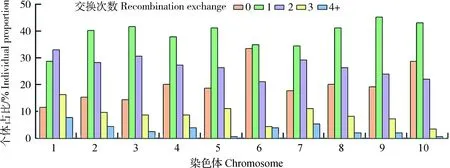
图6 10条染色体中发生不同交换重组次数的个体占比Fig.6 Proportion of individuals with different number of recombination exchanges on 10 chromosomes
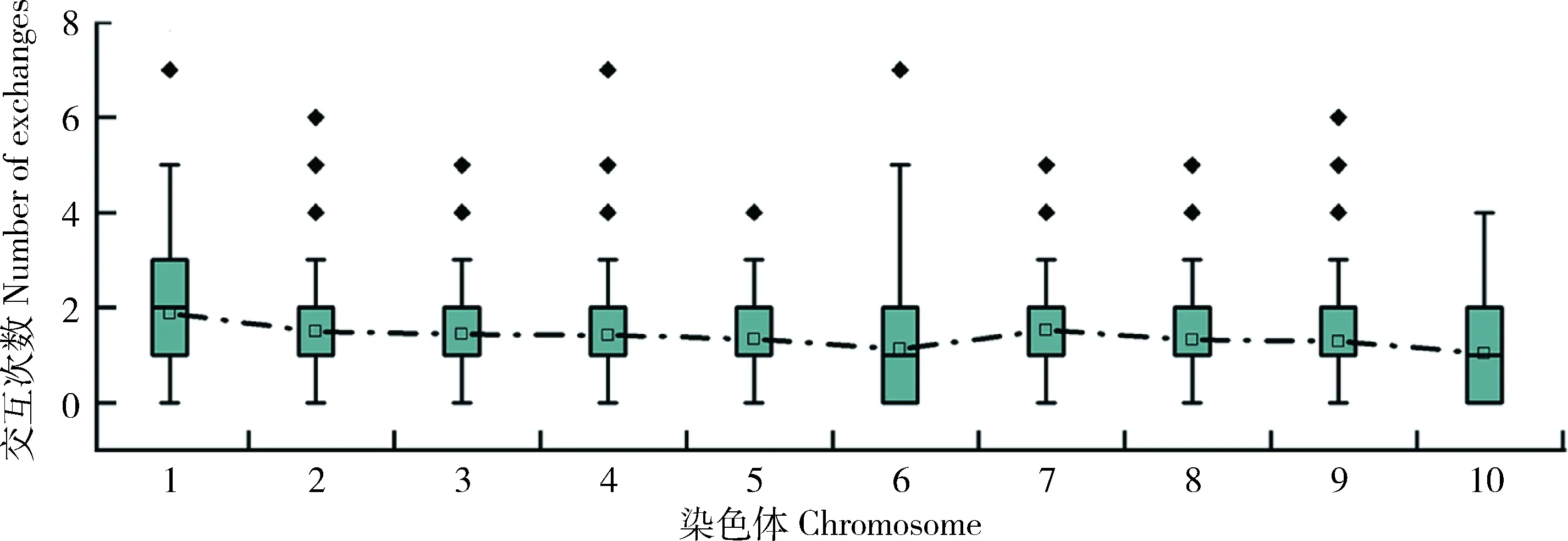
图7 10条染色体重组交换次数Fig.7 Number of recombination exchanges of 10 chromosomes
由图8可知,该DH群体重组交换主要特征包括:1)染色体端粒区域主要为高频交换区,但6号染色体短臂端粒区域与其他染色体端粒区域相比重组交换频率较低。2)1~6号染色体的长臂较长,交换频率从着丝粒到端粒呈现“先上升,再下降,再上升”的特点,但4号染色体的长臂区域存在一个低频交换区。3)根据10条染色体上的交换点位置和交换频率分为2种模式:一是1号和6号染色体出现大量的低频交换点;二是3和8号染色体出现少量高频交换点。
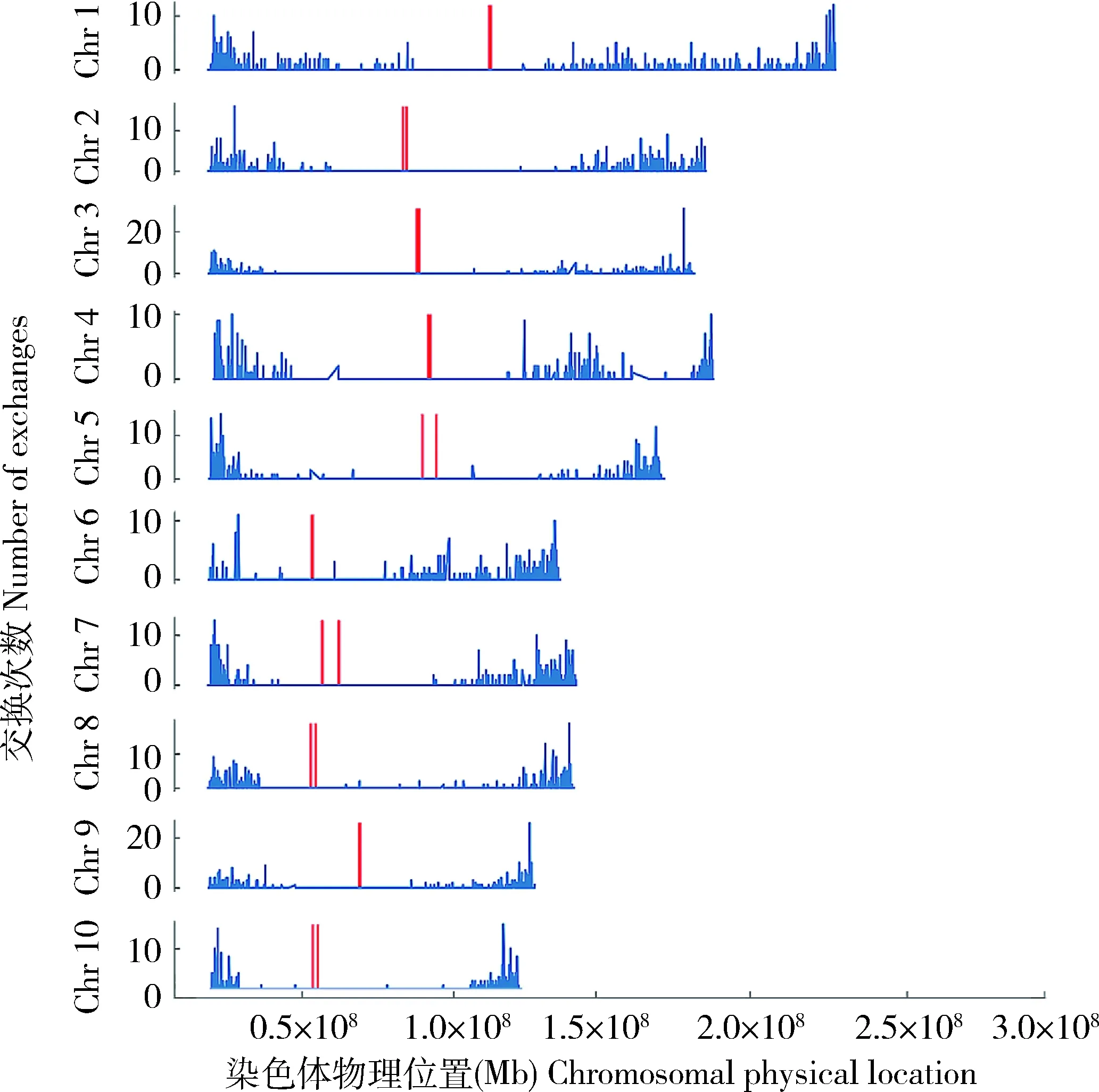
红色线代表染色体着丝粒区域。 The red line represents the centromere region of the chromosome.图8 全基因组重组交换点位置及次数Fig.8 Location and number of genome-wide recombination exchange points
2.4 ‘先玉335’DH群体的分离规律
利用精准分型的高密度分子标记对‘先玉335’DH群体的分离情况进行解析,10条染色体的平均分离系数为51.7%。根据SNP标记在基因组上的分离情况将染色体分为偏母本型、偏父本型和中间型3种类型,见图9。其中1和2号染色体表现为偏母本型,3和8号染色体为偏父本型,4、5、6、7、9和10号染色体为中间型。1号染色体偏向母本的标记数达到1 465个(P
<0.05),3号染色体偏向父本的标记数达到1 439个(P
<0.05)(表2)。在10条染色体着丝粒附近区域,由于发生低频次、低密度的交换,分离曲线变化较为平缓。在非着丝粒区域存在明显的分离拐点,该区域分离曲线剧烈变化且由于出现高频次、高密度的交换,有接近50%理论分离系数的趋势。3 讨 论
3.1 标记筛选模式的优势
本研究采用的标记强筛选模式可发现AxAS软件分型错误的标记,通过精准分型策略自动化获得大量位点的准确分型,可精准地解析DH群体遗传规律。AxAS软件导致分型结果错误的原因主要有2个:一是软件在分型过程中没有预先设置参照,对于基因型出现的位置没有预期判断;二是使用软件预设的自交系分析参数16,默认将基因型分成两个纯合聚类,当数据出现缺失或者杂合基因型时,软件很难识别并且出现聚类紊乱的问题。分型错误会导致后续遗传分析无法获得准确的结果:第一,标记中的杂合分型或缺失分型被统计为纯合分型时,会导致纯合基因型统计错误,统计交换次数和标记分离系数出现偏差。第二,精准分型策略使群体分型准确性大幅提高,群体分离结果中出现的拐点等规律为真实数据的呈现,绘制的分离曲线更加平滑。第三,当分型数据表现非预期规律的现象时,无法对大量标记进行人工校对。而选择直接删除标记,会导致标记数量减少,获得信息不全面。
宋伟等使用Maize SNP3072芯片对回交群体进行分子辅助背景选择,共发现104个标记在双亲间存在多态性,筛选的双亲互补位点数量十分有限。因此使用高质量、高密度的基因芯片能够保证筛选足够的双亲互补标记用于后续研究分析,尤其对于近等基因系、派生品种等背景高度近似的样品可提供更多的信息。本研究使用的Maize6H-60K芯片基数大、密度高,经过强筛选模式仍获得高质量、高密度、双亲互补的可用位点,SNP标记在全基因组上分布均匀,能够满足后续遗传规律研究。
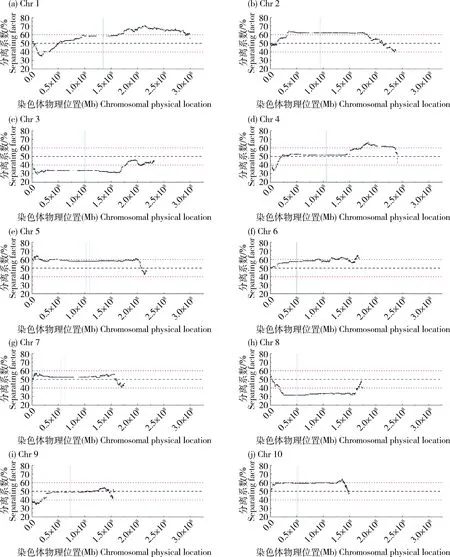
黑色虚线代表理论分离水平,红色虚线代表卡方检验显著水平P=0.05,竖直线代表染色体的着丝粒区域。 The black dashed line represents the theoretical separation level, the red dashed line represents the chi-square test significance level P=0.05, and the vertical line represents the centromeric region of the chromosome.图9 DH群体染色体分离曲线Fig.9 Chromosome separation curve of DH population
3.2 精准分型策略的应用价值
错误的基因分型会在作物遗传改良中带来严重的后果,因此在遗传和育种研究中需要高度重视采集数据后的精准分型问题。本研究发现在使用AxAS软件默认的自交系参数时,会将数据分为2种纯合聚类,可能出现杂合和缺失数据聚类错误。
表2 SNP标记分离情况统计
Table 2 Statistics of separation of SNP markers
染色体Chromosome偏母本型Toward female parent偏父本型Toward male parent中间型Intermediate type位点数量Numberof loci位点占比/%Percentof loci位点数量Numberof loci位点占比/%Percentof loci位点数量Numberof loci位点占比/%Percentof lociChr 11 465.049.8178.06.11 297.044.1Chr 21 268.055.222.01.01 006.043.8Chr 346.02.11 439.064.5746.033.4Chr 4722.035.2117.05.71 210.059.1Chr 5319.014.319.00.91 887.084.8Chr 6491.031.41.00.11 072.068.5Chr 741.02.60.00.01 532.097.4Chr 815.01.01 145.077.4320.021.6Chr 92.00.1239.015.71 277.084.1Chr 10188.03.90.00.01 168.086.1
注:加粗文本表示染色体偏离方向。
Note:The text in bold indicates that the chromosome is segregation distortion.
DH群体便具有背景高度纯合的特点,较少出现杂合基因型,因此可以准确判断位点基因分型结果是否准确。由于在制备群体过程中串粉、机械混杂等因素,以及仍然存在的剩余变异,个别位点不可避免会出现杂合基因型,因此在本研究中删除出现杂合的位点以减少对后续计算分离系数的干扰。
对测序数据和芯片数据的填充算法已有研究结果表明,SNP在基因组上覆盖度较高的前提下,基因型填充的错误率主要取决于SNP分型错误率,但关于降低SNP分型错误率鲜有报道。本研究结果表明AxAS软件分型将缺失或者杂合基因型误判为纯合基因型,影响SNP芯片原始数据的准确率,加大了数据填充的难度,进一步导致对群体结构分析的偏差。当出现基因型错误数据时,软件并不会自动识别矫正,相反在使用单倍型进行填充的过程中还会误导对下游基因型的推测,进而放大基因型错误。本研究使用的精准分型策略能够有效防止出现DH群体分型错误,降低后续基因型芯片数据填充的难度。
本研究在DH群体上首先探索SNP标记精准分型策略的可行性,玉米自交系与DH群体的基因型主要为纯合和少量杂合,同样可以使用精准分型策略以提高自交系的分型准确性和分析精度。当对回交群体、玉米杂交种等材料分型时,由于新增了杂合基因型,精准分型策略还需进一步测试与完善。精准分型策略还有望为其他高通量分型平台解决基因分型问题提供思路。基因芯片平台和KASP平台都通过原始荧光信号值聚类分型,并转化为二维可视图像。但KASP平台通常使用少量标记对大量样品进行基因分型,因此SNP分型错误率对KASP结果的影响更大。根据精准分型策略,在KASP平台基因分型过程中参考历史分型数据有望改善基因分型结果。
3.3 DH群体的遗传规律分析及应用
本研究结果发现,随着染色体长度的减少,染色体交换频率降低,因此平均交换次数也不断降低。6号染色体的交换点数量少、短臂端粒区域与重组交换效率偏低、染色体完整度最高均说明该区段具有特殊结构,杨秀燕等研究表明玉米6号染色体短臂存在随体结构,随体被认为是高度惰性的异染色质,基本没有功能基因的存在。通过掌握群体重组交换情况,能够全面解析DH群体基因组变异规律,从基因组水平上获得目标区段变异个体,并且能够有效控制群体规模,筛选预期材料进行田间测配。
在玉米中,F、RIL等群体中被报道出现偏分离现象,并且主要与配子体基因有关,已有研究证实配子体基因与偏分离热点区域有部分重叠。通过DH群体分离曲线说明,诱导系诱导且自然加倍的玉米DH群体在个别染色体上存在明显的偏分离现象,而刘玉强等研究报道化学加倍的DH群体没有出现偏分离现象,因此,加倍方式也是影响偏分离的因素之一。其他原因还包括配子和合子的选择、母质效应、群体类型、环境因素、标记类型和基因分型错误等。该DH群体出现偏分离现象可能经过环境及非环境因素对无害等位基因双重筛选导致。单倍体植株对田间环境耐受性低,非理想环境对双亲不同的适应能力进行选择,因此表现出染色体整体偏向一个亲本,且在不同染色体上偏向不同的亲本。染色体的大片段偏离与群体田间表型性状不存在高度的相关性,因为株高、容重等性状都是多基因控制,并未受到环境选择,所以田间表型并没有在群体内表现出偏分离。另外值得注意的是偏分离回归现象,即端粒区域的曲线总有回归到理论分离水平的趋势。偏分离回归现象与端粒区域交换频率显著高于着丝粒区域相关,猜测高频交换点为偏分离的回归提供了可能,需进一步试验探究。
目前,永久性遗传群体主要包括DH和RIL群体,相较于F等临时性群体更加适合完成多年限、多试验点的重复研究。DH技术能够最大程度缩短育种时间,降低育种成本,目前已被广泛应用于大规模快速生产自交系。在构建DH群体进行遗传分析时,可以根据初步定位信息设计不同的遗传群体规模。若目标区域初步定位在端粒高交换区域,则使用较小的群体规模即可获得交换单株;如果想获得低交换区的变异,就需要扩大群体规模,并且结合高密度标记筛选跟踪目标区域是否发生交换。配合使用Maize6H-60K等高密度芯片进行DH群体背景扫描,以规避群体配制过程中存在的偏分离的问题,明确群体分离特征并合理使用。
4 结 论
通过结合Maize6H-60K芯片和精准分型策略能够实现玉米DH群体基因型数据的高质量采集。该DH群体10条染色体平均交换次数范围为1.84~1.03次,10条染色体着丝粒和6号染色体的随体等异染色质区域的重组交换点个数和频率明显减少。玉米DH群体通过诱导系诱导且自然加倍后,1、2、3、8号染色体存在明显的偏分离现象。10条染色体的分离曲线是连续的,分离曲线在端粒区域有回归到理想分离水平的趋势。利用获得的重组交换及分离规律可用于控制田间育种提高从DH群体中筛选到预期育种材料的准确性。
