DNA复制调控与肿瘤发生发展的研究进展
2022-10-19王艳珍
王艳珍,陈 亮
(武汉大学生命科学学院,湖北 武汉 430072)
肿瘤是一种生长不受控制的组织,不同类型的肿瘤机理差异很大,但几乎所有的肿瘤细胞都具有若干共同特征,包括持续增殖、防止细胞凋亡和基因组不稳定等[1]。DNA复制调控异常和DNA损伤会造成复制压(replication stress),是基因组不稳定的来源,也是癌前细胞和癌细胞的重要特征[2]。相较于正常细胞,肿瘤细胞在DNA的复制过程中会出现复制起始(replication origin)紊乱、S期检查点(checkpoint)异常、复制叉暂停(replication fork stalling)等现象[2],并具有增殖速度快、细胞周期短、复制潜力无限等特征。因此,靶向DNA复制调控抑制肿瘤细胞增殖成为近年来肿瘤治疗的重要手段,多种相关小分子药物已进行临床试验并取得了较好的研究成果。作者在简单介绍DNA复制及其调控机制的基础上,阐述DNA复制调控在肿瘤发生发展中的重要作用,并对DNA复制调控在肿瘤治疗新方法研究中的作用进行探究,以期为肿瘤治疗提供新方向。
1 DNA复制及其调控机制
完整且准确的DNA复制过程是细胞增殖和维持基因组稳定性的重要基础,任何延迟、阻碍或终止DNA复制的缺陷均会导致DNA复制异常。因此,DNA复制调控的每个阶段都极为重要,其对防止遗传改变的积累、维持细胞生存和组织健康具有重要意义。
1.1 DNA复制
基因组DNA复制分为起始、延伸与终止3个过程。DNA复制起始的基因组区域称为复制起始位点,该区域能被特定蛋白质识别并结合形成复制前复合物(pre-replication complex,pre-RC),即为复制起始的许可(replication origin licensing),其被激活后称为复制起始的起火(replication origin firing)。在G1期,由复制起始识别复合物(origin recognition complex,ORC)亚基ORC1~6形成的复合体被招募组装到复制起始位点,然后募集ATP酶和CDC6并与之结合,进而将DNA复制因子CDT1加载到复制起始位点,进一步共同募集微型染色体维持(MCM)复合物以形成pre-RC[3]。在G1期向S期转变时,DDK和细胞周期蛋白依赖性激酶(cyclin-dependent kinases,CDKs)磷酸化多种复制因子MCM10、CDC45、treslin、GINS、TOPBP1和DNA聚合酶ε(pol ε)以促进它们募集到复制起始位点上,并直接磷酸化MCM2~7复合物,形成CDC45·MCM2~7·GINS(CMG) 复合物,激活MCM复合物的解旋酶活性,从而完成pre-IC的形成[3-4]。进入S期后,MCM复合物诱导其它复制激活相关蛋白质如复制因子C(replication factor C,RFC)、增殖细胞核抗原(proliferating cell nuclear antigen,PCNA)、复制蛋白A(replication protein A,RPA)和其它DNA聚合酶的募集,将pre-IC转化为2个含有复制体的复制叉,复制叉从激活的起点向相反方向移动完成复制起始位点的激活[3]。真核生物中参与DNA复制起始的因子列于表1。
复制起始激活后,MCM10和酸性核质DNA结合蛋白1(acidic nucleoplasmic DNA-binding protein 1,AND-1)募集DNA聚合酶α引物酶(pol α primase),催化合成短的核糖核酸(RNA)引物,引导DNA合成;随着CMG解旋酶向相反方向移动,2个复制叉与polα引物酶、pol δ、pol ε和PCNA结合以促进DNA合成[5];最后,当2个相邻的复制叉从相反的方向相互接近时,拓扑异构酶Ⅰ(topoisomerase Ⅰ,Topo Ⅰ)和TopoⅡ会相继释放扭转压力,复制叉之间的所有蛋白组织(包括核小体)被移除,当最后一个冈崎片段的双链DNA合成并连接后便完成DNA的复制终止[6]。
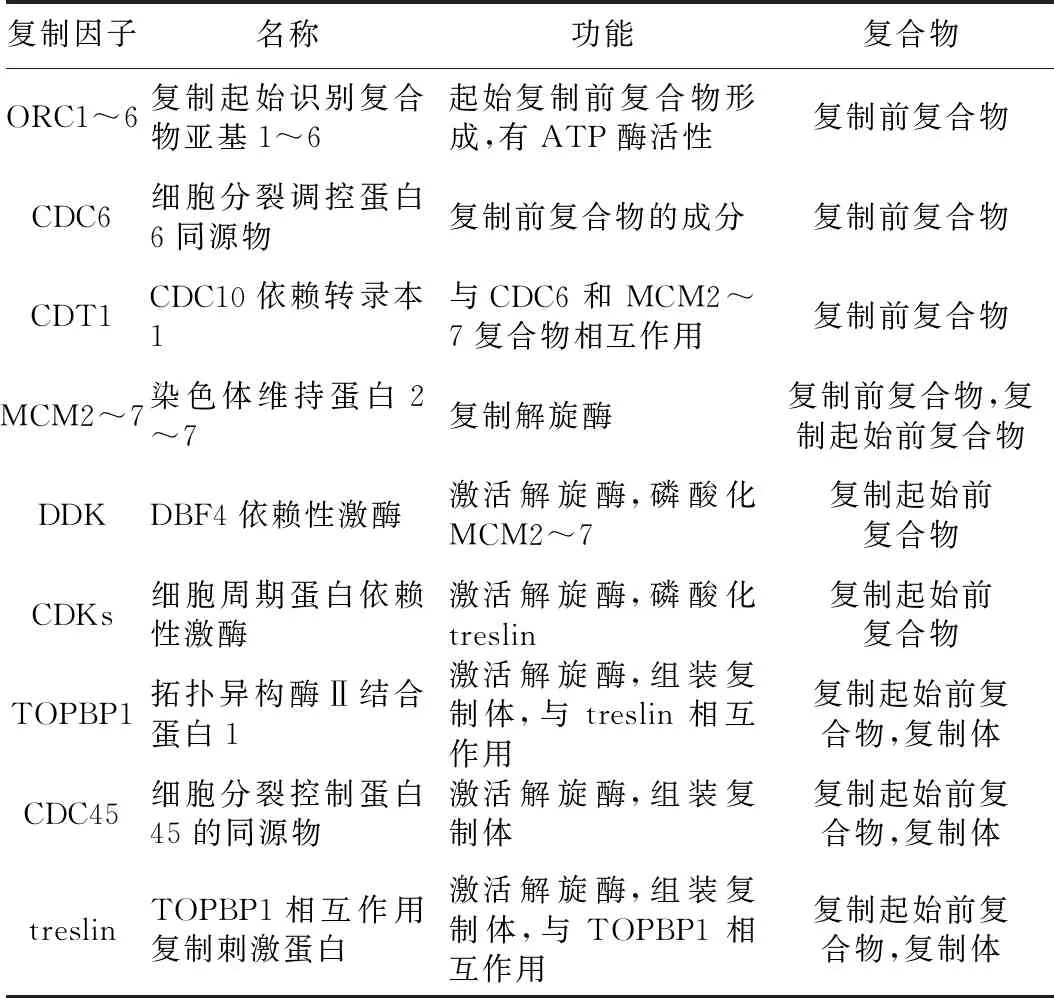
表1 真核生物中参与DNA复制起始的因子
1.2 DNA复制调控机制
DNA复制过程由众多调控因子相互作用共同维持,包括细胞周期及其检查点调控、DNA复制与转录协同调控、多聚ADP-核糖基化修饰机制等。
1.2.1 细胞周期及其检查点调控
细胞周期蛋白(cyclin)、CDKs和CDK抑制因子(CDK inhibitors,CKIs)是参与细胞周期调控的主要因子。CDKs与不同周期表达的细胞周期蛋白相结合,其中CDK4是G1期重要分子,细胞周期蛋白D1能将其激活,导致细胞从G1期加速进入S期。林宇静等[7]发现,在瘢痕癌中细胞周期蛋白D1和CDK4的表达量升高,导致细胞快速增殖,从而诱发癌症。在G2晚期和M早期,细胞周期蛋白A与CDK1结合后使细胞向M期进入。CKIs会抑制细胞周期蛋白与CDKs结合,导致细胞周期无法正常运行。因此,细胞周期蛋白、CDKs、CKIs之间的相互协调是保证细胞周期正常运行的关键。
细胞周期检查点是监测细胞周期主要事件的顺序、完整性和保真度的机制。在检测到DNA损伤后,毛细血管扩张性共济失调突变基因(ATM)和ATM/Rad3相关蛋白基因(ATR)可以将信号传导至整个通路的核心——CHK1;ATR磷酸化激活CHK1,后者磷酸化CDC25,形成ATR-CHK1-CDC25通路,加速CDC25的泛素化降解,最终使细胞周期停滞[8];ATM亦可以通过磷酸化CHK1的Ser317位点激活整个信号通路[9]。在发生DNA双链断裂损伤(double-strand breaks,DSBs)时,ATM-CHK2-CDC25信号通路激活不同的细胞周期检查点,对DNA损伤进行修复,使细胞周期进程发生阻滞[10-11]。
1.2.2 DNA复制与转录协同调控
DNA的复制和转录都是细胞内高频的生物学过程,二者共用DNA模板,并且早期DNA复制和转录都发生在活性染色质区域,可能导致转录-复制冲突(transcription-replication conflicts,TRCs),破坏基因组稳定性和细胞活性,是癌细胞的标志[2]。细胞如何调控转录-复制冲突仍是待阐明的生物学问题,清楚的是细胞依赖多种机制避免或解决冲突。
复制叉的移动方向和转录的延伸方向一致所造成的冲突称为同向(co-directional,CD)的转录-复制冲突,反之称为反向(head-on,HO) 的转录-复制冲突[12],其中,HO-TRCs对细胞危害更大,会造成染色体缺失、重组和细胞死亡[13]。为减少HO-TRCs,细菌基因组倾向于将一些必要且高频转录的基因与复制同向进行[14]。在哺乳动物细胞中,早期复制起始发生在开放的染色质区域并且与转录延伸相互排斥,中频转录的基因组区域通常与高复制起始频率相关,而在高频转录的基因组区域中检测到的复制起始事件非常少[13-14]。复制起始因子激活之前在染色质上的重新分布是减少HO-TRCs的另一种机制,在果蝇中,MCM复合物的基因组分布受转录调控,在基因非转录区域富集,但在转录活跃区域减少[15]。RNA聚合酶Ⅱ促进MCM复合物重新分配在非转录区域,避免转录区域早期DNA复制的起始[14]。Gros等[15]发现,在酵母中, G1期加载在转录终止上的MCM复合物在终止位置突变后被RNA聚合酶重新定位到新转录区域之外。在S期,真核生物将基因组中复制区划分出来,因此,DNA复制与转录协同调控机制发生在DNA复制过程的不同区域、不同时间[16]。
1.2.3 多聚ADP-核糖基化修饰机制
多聚ADP-核糖基化是由多聚ADP-核糖聚合酶(PARPs)催化和转移的动态可逆翻译后修饰。PARP1是PARPs家族主要成员,负责约90%多聚ADP-核糖基(poly-ADP-ribose,PAR)的修饰[17]。PARPs介导烟酰胺腺嘌呤二核苷酸(NAD+)中的ADP-核糖基(ADP-ribose,ADPR)以单个或多个形式与底物蛋白质的特定氨基酸残基发生共价连接反应形成单ADP-核糖基化(mono ADP-ribosylation,MARylation)和多聚ADP-核糖基化(poly-ADP-ribosylation,PARylation)[18]。PAR修饰调控细胞内多种生物学过程,包括复制、DNA修复、转录、代谢、应激和免疫反应等[19-20]。
研究报道,PARPs活性在复制叉处[21]和新复制的染色质区域中[22]增强。PARP1与参与DNA复制的多种蛋白质如pol α、pol δ、解旋酶、拓扑异构酶和PCNA相互作用并刺激其活性[23-24],且这些蛋白很多被PAR修饰,表明PARP1可能作为传感器调控复制叉进程,或通过直接与复制蛋白结合催化其形成PAR来调控[23]。Hanzlikova等[24]发现,S期DNA复制位点检测到的PAR信号在未连接的冈崎片段产生,可能作为单链断裂修复的“备用”机制,促进冈崎片段连接。PARP1还可调控复制压下DNA复制叉的延伸速率,抑制PARP1可反转小分子药物对复制叉进程的抑制[25-26]。在未受干扰的细胞中,PARPs的抑制或敲除不仅导致复制叉延伸速率的加快,并检测到复制起始之间的距离增大[27],进一步验证PAR修饰对复制叉和复制起始的调控作用。
另外,ADP-核糖基化活性也有助于PARP1修复DNA损伤和维持基因组稳定性。作为DNA损伤应答(DNA damage response,DDR)中的信号传感器,PARP1与DNA单链断裂 (single-stranded DNA breaks,SSBs)、DNA缺口或DSBs结合,发生结构变化并激活其催化功能,在自身和邻近蛋白进行PAR修饰,促进如X-线修复交叉互补基因1(X-ray repair cross complementing group1,XRCC1) 等DNA修复因子的募集,改变染色质结构并促进DNA损伤修复[28]。由于PAR修饰在多种DNA损伤中的修复作用,PARPs抑制剂的潜在抗肿瘤作用逐步被揭示并在临床治疗中被应用。
2 DNA复制调控在肿瘤发生发展中的重要作用
2.1 肿瘤的特点与形成原因
人类肿瘤形成是一个多步发展的过程[29],受物理、化学、生物等因素诱导。基因组不稳定性是驱动肿瘤发展并获得基因突变和选择性生长的重要因素[30]。例如某些环境化合物分子与DNA形成加合物,有可能造成特定肿瘤抑制基因或原癌基因突变和DNA复制压,促使细胞癌化并持续增殖。
2.2 DNA复制在肿瘤发生发展中的重要作用
DNA复制异常体现在持续复制、复制叉停滞和崩溃、DNA损伤及DDR异常、检查点和细胞周期失活、DNA损伤修复错误、复制重启错误等。这些异常是推动肿瘤发展和基因组不稳定的重要因素。肿瘤细胞具有无限的复制潜力,并规避复制异常缺陷。例如细胞周期调控机制在人类肿瘤中经常失调,导致肿瘤细胞周期蛋白的异常激活。研究[31]表明,失调的CDKs诱导非计划性增殖和基因组不稳定性,DNA损伤和有丝分裂检查点的改变经常导致CDKs活性增强,从而驱动肿瘤细胞周期进程。核心细胞周期机制内的遗传损伤导致其过度激活,在大多数肿瘤的发生发展中起着重要作用,并且肿瘤细胞中组成性有丝分裂信号传导和对抗有丝分裂信号的缺陷反应导致细胞恶性增殖[32-33]。
DNA损伤修复机制在肿瘤发生发展中也起着关键作用。鉴于肿瘤细胞形成多种DNA损伤及基因组不稳定性,其生存更依赖DDR途径。但与正常细胞不同的是,大多数肿瘤细胞会失去一个或多个DDR途径或能力,从而导致对剩余途径的更大依赖[34]。近年来,肿瘤的靶向治疗被更多地应用到临床,更多靶向DNA复制的小分子药物也被应用到临床。
3 DNA复制调控在肿瘤治疗新方法研究中的作用
3.1 肿瘤的靶向治疗
分子生物学和肿瘤生物学的进步极大地改变了肿瘤治疗方式,从传统的手术、放疗、化疗到靶向治疗、免疫治疗和微创治疗等,不断涌现出新的治疗方法。其中分子靶向治疗是一种利用靶向特定致病分子的药物进行治疗的手段,具有快速有效和便于推广的优势。理想靶点的识别对于靶向疗法的成功开发至关重要,肿瘤细胞特异性基因/蛋白是常见靶标[35]。通过了解肿瘤特定分子靶点的生理学特征,可以确定抑制肿瘤发生发展的潜在分子策略。复制起始异常、DNA损伤、肿瘤特异性DNA修复缺陷等正常细胞的差异性特征,为肿瘤的分子靶向治疗提供了新方向。
3.2 利用DNA复制起始开发肿瘤治疗新方法
靶向DNA复制起始是肿瘤治疗的重要方向。DNA复制起始蛋白常在肿瘤细胞中高表达,而在非增殖正常细胞中不表达或低表达。因此,DNA复制起始蛋白可能成为肿瘤治疗的潜在靶点,如靶向细胞分裂周期7(cell division cycle 7,CDC7)激酶[36-39]可有效且特异性杀伤肿瘤细胞,其作用机制为:在抑制CDC7后,正常细胞能够利用细胞周期检查点将细胞停滞在G1/S期交界处并暂停DNA复制,而肿瘤细胞由于缺失细胞周期检查点依赖的基因,进入S期后没有足够复制起始位点的激活而造成DNA复制异常,最终造成肿瘤细胞死亡。目前,多家生物制药公司已启动CDC7药物开发计划并进入早期临床试验[40-41]。Feng等[42]运用靶向3个人类DNA复制起始基因hCdc6、hMcm2和hCdc45的反义寡核苷酸和siRNA分子,显著降低靶基因产物的mRNA和蛋白质水平,并阻止了DNA复制和细胞增殖,导致p53阳性和阴性肿瘤细胞凋亡,而不会导致正常细胞死亡。另有研究表明,抑制复制起始蛋白CDT1p活性的双核蛋白片段可以抑制DNA复制和细胞增殖,并导致肿瘤细胞凋亡[43],siRNA对ORC6p(起始蛋白ORC的一个亚基)的基因沉默导致细胞周期失调,包括有丝分裂阻滞和多核细胞出现[44],进一步证明抑制复制起始蛋白活性的小分子是潜在的抗肿瘤药物。
3.3 利用DNA复制应激开发肿瘤治疗新方法
DNA复制应激是导致肿瘤基因组不稳定的主要因素之一[45-46]。许多肿瘤抑制基因(tumor suppressor gene,TSG)的突变都会引发DNA复制应激并激活ATR、CHK1和酪氨酸激酶WEE1等以确保复制叉的稳定和修复,并防止异常复制的基因组进入有丝分裂M期[47-48]。因此,抑制DNA复制应激和修复DNA损伤是提高肿瘤治疗效果的途径之一。
很多肿瘤细胞G1检查点失调,使其依赖于由ATR-CHK1通路调控的S/G2检查点。除调节细胞周期检查点外,ATR-CHK1在复制应激时能够保护细胞免受DNA损伤、稳定停滞的复制叉、防止其崩溃和DSB形成、控制补偿性起始位点的激活并促进同源重组[49-50],这些特性使其成为治疗干预的潜在目标。目前,靶向ATR和CHK1的抑制剂已经分别进入临床Ⅱ期和Ⅰ期。DNA复制应激时CHK1还可以激活WEE1,使其抑制CDK1,从而将细胞阻滞在G2/M过渡期,靶向WEE1的抑制剂AZD1775也已进入临床Ⅱ期[45]。此外,更多在DNA复制应激中起调控作用的蛋白抑制剂也不断涌现,包括TopoⅠ和TopoⅡ抑制剂、烷化剂和铂化合物等。
3.4 PARPi在肿瘤治疗中的应用
PARPs抑制剂(PARPi)是以合成致死的理念被用于BRCA1/2突变的肿瘤治疗[51]。BRCA1和BRCA2是介导DNA双链断裂后同源重组(homologous recombination,HR)修复的关键蛋白,BRCA1/2突变同时抑制PARPs即同时阻断单链修复和HR修复对肿瘤细胞致死,为PARPi的理论基础。此外,PARPs参与并调控许多DNA损伤修复的过程包括DNA单链断裂(SSB)、双链断裂(DSB)、碱基切除修复(BER)和核苷酸切除修复(NER)等[45],而肿瘤细胞更依赖PARPs的修复作用,因此,PARPi在肿瘤治疗方面的应用潜力巨大。
目前,有4种PARPi(奥拉帕利、卢卡帕利、尼拉帕利和他拉唑帕利)已获得美国食品药品监督管理局(FDA)批准,主要用于治疗卵巢癌、乳腺癌、前列腺癌、胰腺癌等,还有很多PARPi正处于不同的临床试验阶段,以治疗更多依赖PARPs的肿瘤。PARPi除单药使用外,与化疗或放疗联合亦能提高肿瘤细胞化疗或者放疗的敏感性,并一定程度上克服了耐药性,如PARPi与ATR、血管内皮生长因子受体(vascular endothelial growth factor receptor,VEGFR)、CDKs、WEE1、磷脂酰肌醇3-激酶(phosphatidylinositol-3-kinase,PI3K)等激酶抑制剂联用均表现出显著的协同抗肿瘤作用。PARPi自上市以来显著改善了部分肿瘤的治疗现状,使患者受益。因此,寻找更适合PARPi使用的肿瘤生物标志物、优化PARPi联合治疗方案、克服耐药性以及降低毒副作用已成为肿瘤治疗的研究热点。
4 展望
DNA复制失调是肿瘤基因组不稳定的主要驱动因素,靶向DNA复制的药物成为肿瘤治疗的重要手段。目前这些药物的设计主要是针对肿瘤细胞相对于正常细胞表现出的脆弱性,如肿瘤细胞更加依赖某些DDR通路或DNA复制起始和细胞周期调控的蛋白。因此,找到肿瘤细胞更多的补偿性DDR通路和肿瘤发生发展中的关键性脆弱点将为肿瘤治疗提供更多可能。
