混合型反相微乳体系萃取茶渣蛋白的工艺优化
2022-08-03谭梓铭解新安
张 阳,代 成,谭梓铭,李 璐,李 雁,解新安,2,
(1.华南农业大学食品学院,广东广州 510642;2.广东省功能食品活性物重点实验室,广东广州 510642)
茶叶是一种起源于中国的产品,是一种天然的健康饮料,在世界各地工厂的加工过程中,会产生大量的茶叶残渣[1-2]。据报道,茶渣中含有多种活性成分,其中粗蛋白含量为20%~30%,且80%以上为非水溶性蛋白质[3]。茶渣蛋白由多种氨基酸组成,具有较高的营养价值,此外,它还具有较好的防辐射[4]、抗氧化[5]和降血脂的功能[6],是一种有着极大开发潜力的优质植物蛋白源[7]。目前,茶渣蛋白主要是通过碱提取法[8]和酶提取法[9]来进行萃取。然而,这些方法有着一定的局限性,如环境污染和资源消耗等问题,无法大规模生产。
微乳提取被认为是取代传统方法的理想选择[10-11]。微乳是一个单一的光学各向同性且热力学稳定的体系,是一种含有水相、油相和表面活性剂的自发形成的宏观分散溶液[12-13]。其中常见的微乳为水包油(O/W)型,而油包水(W/O)微乳也称为反相微乳。已经证明了W/O 微乳体系中的“水池”比纯水有更好的溶解能力[14]。因此,W/O 型微乳已被广泛用于植物蛋白的提取[15]。反相微乳萃取茶渣蛋白的过程包括两个步骤:前萃和后萃,其中蛋白质从水相进入表面活性剂聚合物的水池中称为前萃,而含有这些蛋白质的溶液从反胶束被回收到新的水溶液中的过程称之为后萃[16-17]。
已有报道表明,反相微乳提取的植物蛋白具有更好的功能特性和结构[18]。Zhao 等[19]使用AOT 反胶束提取大豆蛋白,发现其具有更好的功能、营养和风味特性,并且FTIR 和氨基酸分析结果表明,大豆蛋白在AOT 反胶束中几乎保持了原有的结构。可能是因为反相微乳中的反胶束结构对植物蛋白有着一定的保护作用[20-21]。此外,Zhu 等[22]研究发现,与碱提法和等电点沉淀法制备的蛋白相比,反胶束法提取的脱脂小麦胚芽蛋白具有相对较好的氮溶性、脂肪吸收能力、起泡性、起泡稳定性和乳化稳定性,结构也更加致密有序。因此使用反相微乳能够从茶渣中提取优质的茶渣蛋白,但目前用反相微乳法从茶叶残渣中提取蛋白质的研究鲜有报道。
在本实验室以前的研究中,Tween80、AOT、SDS和CTAB 四种表面活性剂制成的反相微乳对茶渣蛋白的提取效率不高,因此本文使用Tween80、AOT、SDS 和CTAB 制备了Tween80-AOT、Tween80-CTAB、Tween80-SDS 三种反相微乳体系,并研究了各种因素(前萃:表面活性剂浓度、离子型表面活性剂含量、pH、W0、萃取温度和萃取时间,后萃取:KCl 浓度、缓冲液pH 和提取温度)对反相微乳提取茶渣蛋白的影响。然后通过正交试验对提取条件进行了优化,并通过SDS-PAGE 电泳测定了由反相微乳提取的茶渣蛋白的分子量,为后续生产中进一步开发深加工的茶渣资源提供理论依据。
1 材料与方法
1.1 材料与仪器
茶叶 广州曼陀岭茶厂提供;AOT、SDS、CTAB、KCl 溶液 广州裕兆科技有限公司提供;Tween80、异辛烷、正辛醇 广州光华科技有限公司;一级水实验室自制;所有有机溶剂 均为分析纯。
PL203 电子天平 梅特勒-托利多仪器有限公司;DF-15 型中药粉碎机 温岭市大德中药机械有限公司;KQ-100 超声波清洗机 昆山市超声仪器有限公司;磁力搅拌器 江苏市富华有限公司;HH-2 数显恒温水浴锅 常州澳华仪器有限公司;KJELTEC 8200 快速定氮仪 福斯特卡托公司;DL-5 高速离心机 上海安亭科学仪器厂;FD-EC-80 冷冻干燥机、PHS-3C 数显pH 计 上海精科仪器厂。
1.2 实验方法
1.2.1 茶渣的制备 将蒸馏水加热至沸腾状态,然后将茶叶按料液比1:40 加入并煮沸提取20 min。随后,用棉布将提取物过滤出来。上述处理重复2次后放入烘箱干燥至恒重,然后用DF-15 型中药粉碎机将茶渣磨碎。磨碎的茶渣用60 目网筛过筛[23]。
1.2.2 反相微乳的制备 将一定浓度的表面活性剂溶解在由异辛烷和正辛醇组成的有机溶剂中,得到反相微乳,三种混合表面活性剂(Tween80-AOT、Tween80-CTAB、Tween80-SDS)由两种不同的表面活性剂按一定质量比(w/w)混合组成。然后用磁力搅拌器搅拌混合后的溶液,直到完全溶解。反相微乳体系中的水含量表示为W0(水与表面活性剂的摩尔比),用含有氯化钾的磷酸盐缓冲液调节离子强度和pH。然后,溶液在超声波条件下反应30 min,直到其清晰透明[24]。
1.2.3 蛋白质含量的测定 茶渣蛋白含量的测定参考GB 5009.5-2010《食品安全国家标准食品中蛋白质的测定》中的凯氏定氮法。
1.2.4 蛋白质前萃实验 取按试验要求配制的反相微乳置于锥型瓶中,加入一定量的茶渣,超声一定的时间,然后4000 r/min 离心分离20 min。萃取体系分为两层,上层为萃取蛋白质的反相微乳层,下层为残渣,除去残渣,测量上清液(即为前萃液)体积,取5 mL 上清液定氮(以萃取前的反相微乳作为空白样)[25]。然后以茶渣蛋白前萃率为指标,考察表面活性剂浓度(0.02、0.04、0.06、0.08、0.10、0.12、0.14 g/mL)、离子型表面活性剂质量分数ω(占表面活性剂总质量的比例,10%、20%、30%、40%、50%、60%、70%、80%、90%)、W0(5、10、15、20、25、30、35)、水相pH(7、8、9、10、11、12、13)、萃取温度(25、30、35、40、45、50、55、60 ℃)以及萃取时间(20、40、60、80、100、120 min)5 种因素不同水平对前萃过程的影响。固定考察因素表面活性剂浓度0.08 g/mL,离子型表面活性剂质量分数50%,W025,水相pH10,萃取温度40 ℃,萃取时间60 min。蛋白质前萃率由下式计算:

根据单因素实验的结果,选择了表面活性剂浓度(A)、离子型表面活性剂的质量分数(B)、水相增溶能力W0(E)和水相pH 四个因素。以茶渣蛋白前萃率为指标,通过正交试验确定最佳提取工艺,因素水平设计如表1所示。

表1 前萃正交试验因素与水平L16(45)Table 1 Factors and levels of forward extraction orthogonal test L16(45)
1.2.5 蛋白质后萃取实验 取10 mL 前萃液置于锥形瓶中,加入等体积的具有一定离子强度和pH 的KCl 缓冲液,超声波萃取一定时间,然后4000 r/min离心分离20 min。分液漏斗分层后取下层水相,测量体积,取5 mL 下层水相定氮(以KCl 缓冲液作为空白样),余者真空冷冻干燥,得粗蛋白,测定蛋白质含量[25]。然后以茶渣蛋白后萃率为指标,考察了缓冲液KCl 浓度(0.25、0.5、1.0、1.5、2.0 mol/L)、缓冲液pH(5.5、6.0、6.5、7.0、8.0、9.0、10.0、11.0、12.0)以及萃取温度(25、30、35、40、45、50、55、60 ℃)三个因素不同水平对后萃过程的影响。后萃单因素实验初始条件为:KCl 浓度0.08 mol/L、水相pH7 以及萃取温度40 ℃。蛋白质后萃率由下式计算:

通过正交试验进一步研究了茶渣蛋白后萃的最佳条件。如表2所示,对氯化钾的浓度(A)和pH(B,C)以及提取温度(D)进行考察,每个因素选取三个水平,进行正交优化试验。
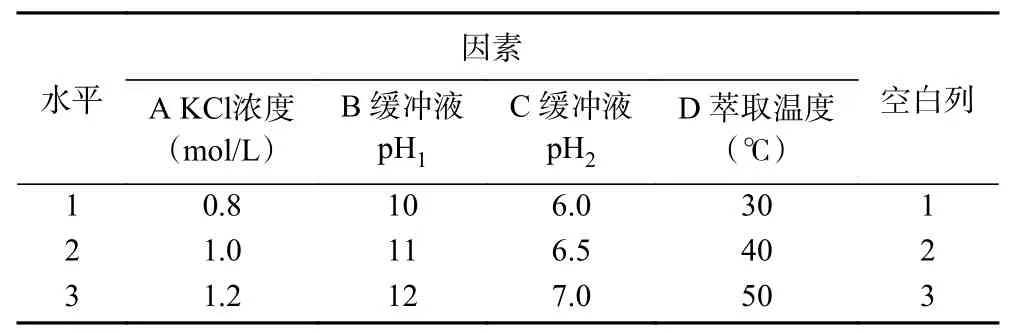
表2 后萃正交试验因素与水平L9(34)Table 2 Factors and levels of forward extraction orthogonal testL9(34)
1.2.6 茶渣蛋白分子量测定 参照何忠效的《生物化学试验技术》中的试验方法。使用4%的浓缩胶和15%的分离凝胶进行SDS-PAGE 电泳分析。含有相同数量的蛋白质的样品与分子量的标记蛋白一起被加载到丙烯酰胺凝胶上。电泳步骤结束后,使用考马斯亮蓝R-250 染色1 h,然后用脱色液(乙醇:乙酸:蒸馏水=25:8:67)脱色2 h。
1.3 数据处理
实验结果为重复三次实验后,取平均值。所有的实验数据都使用SPSS 软件包(11.5 版)进行分析,并使用Excel 2013(Microsoft Corporation,Redmond,WA,USA)绘制了曲线。然后进行方差分析以评估独立之间的显著差异,并通过邓肯多重范围检验(DMRT)评估平均值之间的差异是否显著,不同的小写字母表示具有显著性差异(P<0.05)。
2 结果与分析
2.1 茶渣蛋白前萃实验结果
2.1.1 表面活性剂浓度对蛋白质前萃率的影响 由图1可知,随着表面活性剂浓度的增加,Tween80-SDS 和Tween80-CTAB 反相微乳的前萃率先增加后减少,当表面活性剂浓度为0.10 g/mL 时,前萃率达到最高值。Tween80-AOT 反相微乳体系的前萃率低于上述两种微乳体系,当表面活性剂浓度为0.08 g/mL 时,萃取率达到最大值9.5%。这一结果可能是表面活性剂浓度的影响造成的,表面活性剂的浓度升高后增强了微乳中水的增溶作用,从而增大了胶束的尺寸和/或增加了胶束的数量[26],使蛋白质更有选择性地增加转移到有机相。另一方面,表面活性剂浓度过高,不利于茶渣蛋白的前萃取。因此选择0.08 g/mL 的表面活性剂浓度(Tween80-AOT)和0.10 g/mL 的 表 面 活 性 剂 浓 度(Tween80-SDS 和Tween80-CTAB)进行后续实验。
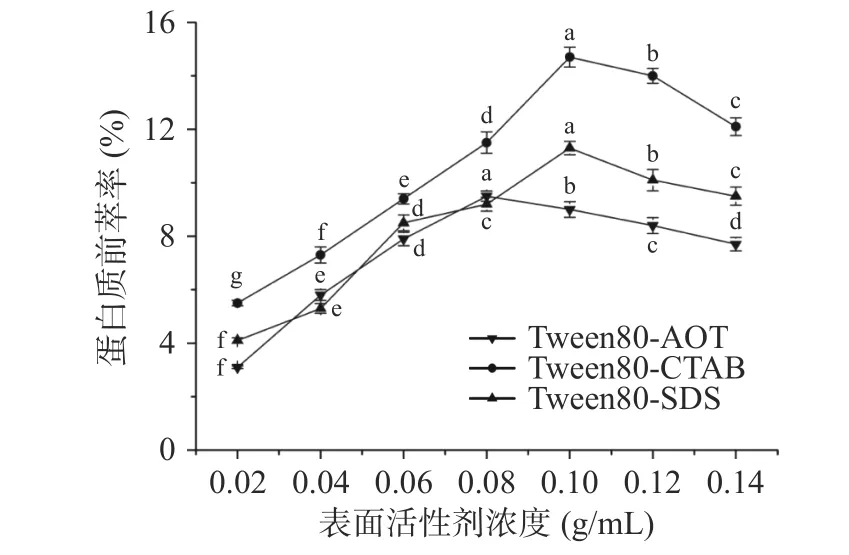
图1 表面活性剂浓度对前萃率的影响Fig.1 Effect of surfactant concentration on the forward extraction
2.1.2 离子型表面活性剂质量分数对蛋白质前萃率的影响 在离子型表面活性剂含量增加的条件下(图2),三种不同的反相微乳体系提取的茶渣蛋白含量均迅速攀升,然后下降。在Tween80-CTAB 和Tween80-SDS 微乳体系中,CTAB 和SDS 含量为70%时,萃取率达到最高,分别为15.1%和11.9%。而在Tween80-AOT 微乳体系中,AOT 含量从10%到80%变化时,前萃率逐渐增加,且AOT 含量增加到80%时,茶渣蛋白的前萃率接近最大值(10.9%),然而,当AOT 含量进一步增加时,前萃率反而开始下降。因此在配制混合反相微乳时,本实验选取AOT 的含量为80%,选取CTAB 与SDS 的含量为70%。
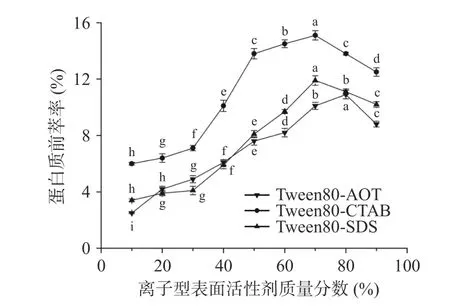
图2 离子型表面活性剂质量分数对前萃率的影响Fig.2 Effect of ionic surfactant content on the forward extraction
2.1.3 W0对蛋白质前萃率的影响 当W0从5 增加到30 时,Tween80-CTAB、Tween80-AOT 和Tween80-SDS 反相微乳体系的前萃率都有极大的提升(图3),当W0达到25 时,前萃率的增加趋势趋于平缓。W0的增加对应于胶束大小的增加,这与蛋白质的溶解度密切相关。这些现象可能是因为随着W0的增加,一些较大的反相胶束形成,可以包含多个蛋白质分子[27]。因此,最佳的W0应该是25。
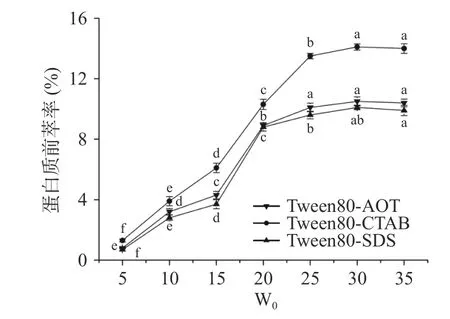
图3 W0 对前萃率的影响Fig.3 Effect of W0 on the forward extraction
2.1.4 水相pH 对蛋白质前萃率的影响 从图4 可以看出,在三种反相微乳体系中中,茶渣蛋白前萃率的变化与pH 的变化趋势不同。对于Tween80-AOT和Tween80-SDS 微乳体系,前萃率先上升然后略有下降,当pH 为9 时达到最大值。对于Tween80-CTAB微乳体系,前萃率随着pH 的增加而增加,但当pH达到12 时,茶渣蛋白的前萃率(14.2%)最高。相比于其他两种反相微乳体系在相同pH 下的提取效果,Tween80-CTAB 微乳体系对茶蛋白的提取率最高。在最佳pH 下,蛋白质的提取率会通过静电相互作用或疏水相互作用得到改善[16]。过高或过低的pH 不仅会导致蛋白质变性,而且由于将蛋白质和表面活性剂混合成乳状液而无法形成反相胶束[28]。后续实验Tween80-AOT 和Tween80-SDS 微乳体系的水相pH选择为9.0,而Tween80-CTAB 微乳体系的水相pH选择12.0。
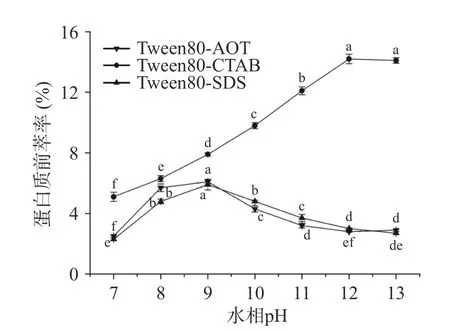
图4 pH 对前萃率的影响Fig.4 Effect of pH on the forward extraction
2.1.5 萃取温度对蛋白质前萃率的影响 如图5所示,随着温度的升高,微乳的整体萃取趋势表现为先升高然后降低。当萃取温度为45 ℃时,Tween80-AOT 反相微乳体系获得了最大的萃取率,而当温度为40 ℃时,Tween80-CTAB 体系和Tween80-SDS体系的萃取率最高,然后当温度继续上升时,萃取率逐渐开始下降。这表明温度过高或过低都不利于蛋白质的前萃取,不恰当的温度会影响蛋白质在微乳中的溶解度[29-30]。而在适当的温度下,蛋白质和反相胶束分子之间的相互作用会增强[31]。因此,本实验中Tween80-AOT 微乳体系的适宜萃取温度为45 ℃,Tween80-SDS 和Tween80-CTAB 微乳体系的适宜提取温度为40 ℃。
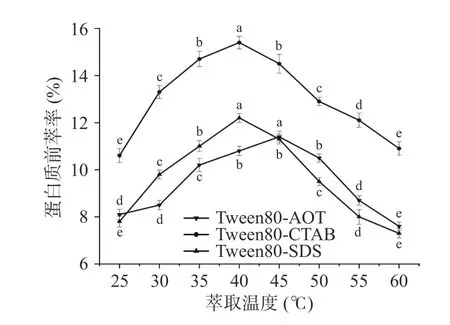
图5 萃取温度对前萃率的影响Fig.5 Effect of temperature on the forward extraction
2.1.6 萃取时间对蛋白质前萃率的影响 在图6 中,前萃率在提取时间为20~60 min 的范围内明显增加,随后在60~120 min 之间的上升趋势趋于平缓。这表明在早期阶段(20~60 min),蛋白质的前萃取容易受到提取时间的影响,而且提取率随着提取时间的增加而上升。在60~80 min 之间,茶渣蛋白的前萃率没有随着提取时间的增加而发生明显的改变,这可能是由于在前期的萃取过程中,反胶束已经增溶了大量的茶渣蛋白,达到一个饱和状态,并且随着萃取时间的增加,离子强度等因素也会对反胶束结构造成一定的影响[31],因此在随后实验中萃取时间选择40 min。
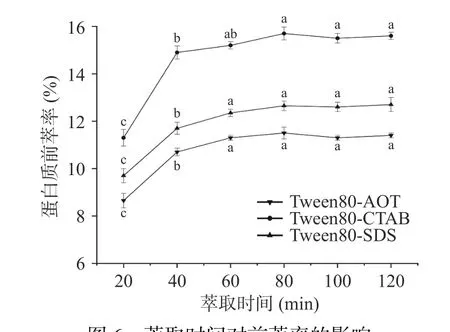
图6 萃取时间对前萃率的影响Fig.6 Effect of extraction time on the forward extraction
2.1.7 前萃工艺的正交试验 根据上述单因素实验的结果,对影响前萃取的四个主要因素(表面活性剂浓度、离子型表面活性剂的质量分数、W0和水相的pH)进行了正交优化试验。从表3 中正交试验结果可以看出,影响Tween80-AOT、Tween80-CTAB 和Tween80-SDS 三种反相微乳体系前萃率的主次顺序分别是W0>pH>表面活性剂浓度>离子型表面活性剂质量分数、表面活性剂浓度>pH>W0>离子型表面活性剂质量分数和表面活性剂浓度>pH>离子型表面活性剂质量分数>W0。且从表4~表6 中可以看出,仅有W0对Tween80-SDS 体系的前萃率影响不显著。实验结果表明,Tween80-AOT 微乳体系中四个因素的最佳组合是:A3B2C4E3,即在表面活性剂浓度为0.10 mol/L、离子型表面活性剂含量为70%、pH 为10.0、W0为25 的条件下,Tween80-AOT 微乳体系的茶渣蛋白前萃率最高(11.49%)。Tween80-CTAB 微乳中四个因素的最佳组合是:A3B2D4E3,茶蛋白的最大前萃率的条件为:表面活性剂浓度0.10 mol/L,离子型表面活性剂含量70%,pH13.0,W025,最高前萃率为16.17%。Tween80-SDS 体系中四个因素的最佳组合是:A2B3C3E3,在最佳条件下(表面活性剂浓度为0.08 mol/L,SDS 含量为80%,pH 9.0,W025)进行实验后,前萃率为13.10%。
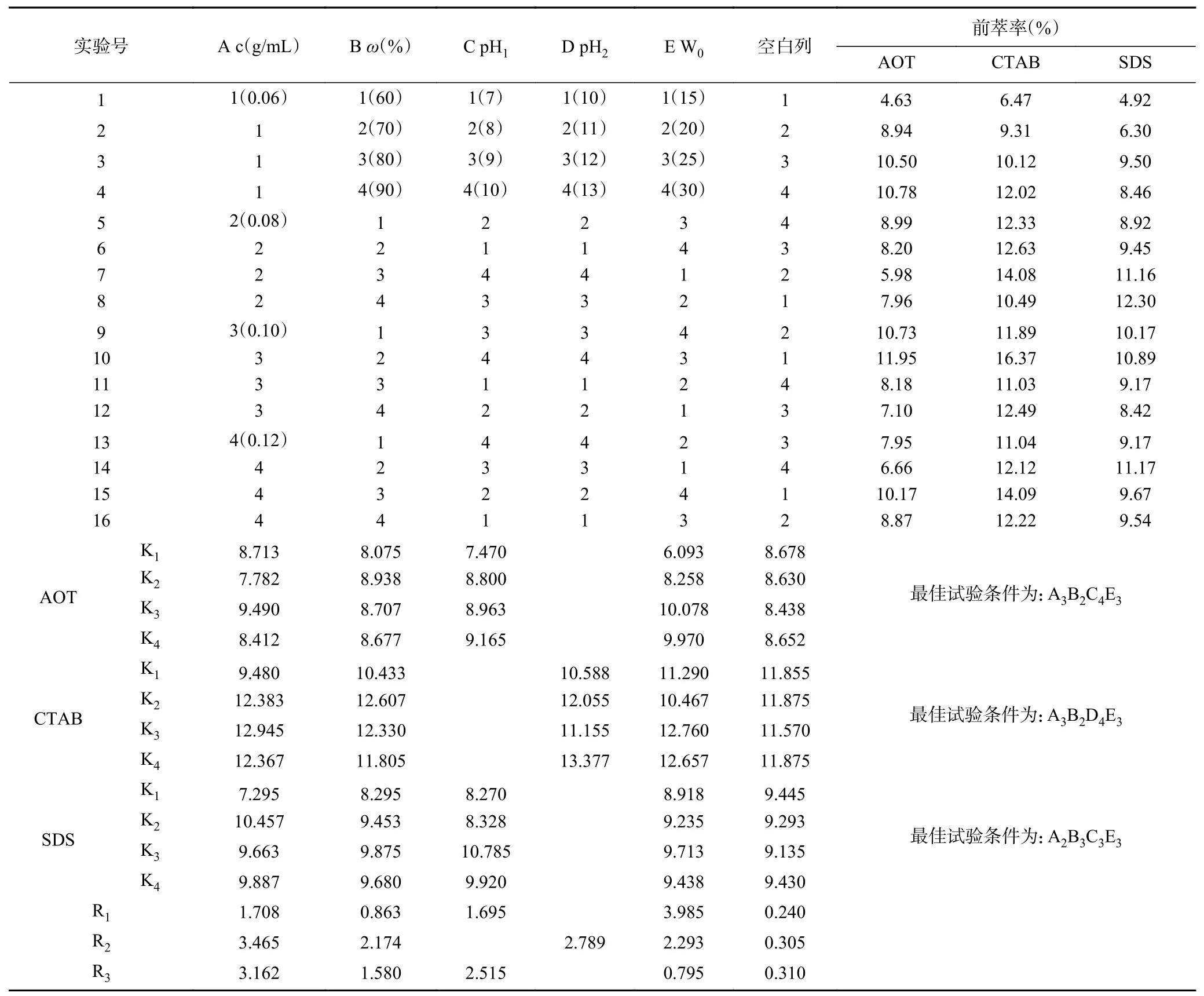
表3 前萃正交试验安排与结果Table 3 Arrangement and results of forward extraction orthogonal experiment

表4 Tween80-AOT 体系前萃正交试验方差分析表Table 4 Significant analysis on forward extraction orthogonal test of Tween80-AOT system

表6 Tween80-SDS 体系前萃正交试验方差分析表Table 6 Significant analysis on forward extraction orthogonal test of Tween80-SDS system

表5 Tween80-CTAB 体系前萃正交试验方差分析表Table 5 Significant analysis on forward extraction orthogonal test of Tween80-CTAB system
2.2 茶渣蛋白后萃实验结果
2.2.1 缓冲液KCl 浓度对蛋白质后萃率的影响 图7显示了KCl 浓度对后萃率的影响。随着KCl 浓度的增加,三种反相微乳体系的后萃率均表现为先上升后下降,当KCl 浓度为1.0 mol/L 时达到最大值,其中Tween80-CTAB 最大后萃率为94.5%,而Tween80-AOT 和Tween80-SDS 最大后萃率分别为90.21%和89.41%。研究发现,随着KCl 浓度的增加,表面活性剂周围的电层厚度逐渐变薄,表面活性剂的极性头之间的相互排斥力降低,导致反相微乳中的胶束变小,使得微乳中蛋白质的增溶作用减弱,后萃率得到提高[32]。
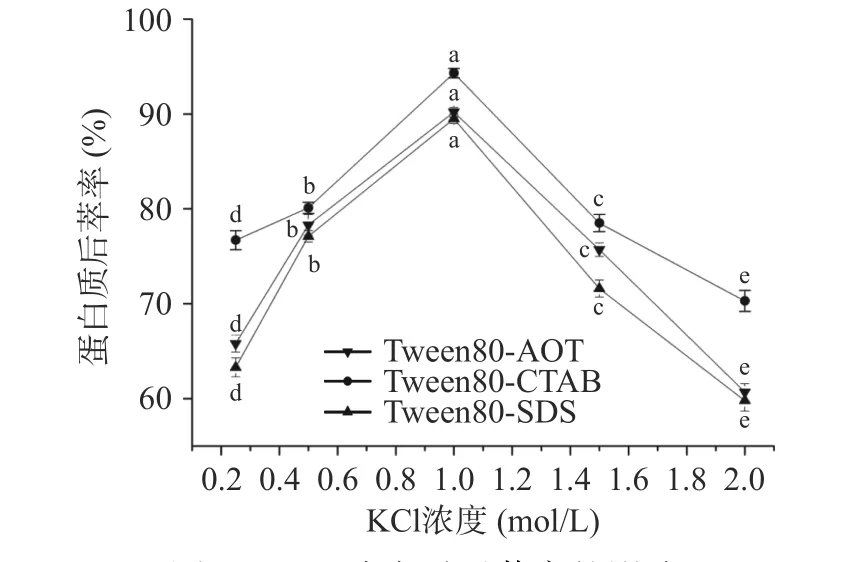
图7 KCl 浓度对后萃率的影响Fig.7 Effect of KCl concentration on the backward extraction
2.2.2 缓冲液pH 对蛋白质后萃率的影响 如图8所示,Tween80-AOT 和Tween80-SDS 微乳体系的茶渣蛋白后萃率随着pH 的升高而增大,当pH 在5.5~11 时,茶渣蛋白的后萃率明显上升,在pH 到达11 后,出现缓慢下降的趋势,因此在pH 为11 时达到最高值。在Tween80-CTAB 体系中,pH 在5.5~6.5时,蛋白质的后萃率大大增加,然后随着pH 的增加逐渐降低,茶渣蛋白的后萃率在pH 6.5 时达到最高。缓冲液pH 影响后萃率的原因是随着pH 的增加,更多的茶渣蛋白分子带负电,电荷密度增加,削弱了蛋白质与表面活性剂之间的静电作用,促进了后萃取过程,即蛋白质从反相胶束中的释放率增加[33]。另一方面,较高的pH 反而会增大蛋白质之间的静电排斥作用,导致蛋白质后萃率下降[34]。

图8 缓冲液pH 对后萃率的影响Fig.8 Effect of buffer pH on the backward extraction
2.2.3 萃取温度对蛋白质后萃率的影响 反相微乳中茶蛋白的提取率可能受到温度的影响[24]。从图9中可以得知,三种反相微乳体系对茶渣蛋白的提取率趋势一致,且在40 ℃时后萃率达到最高值,但随着温度的持续升高,后萃率均明显下降。原因是在温度升高之初,反相微乳与水相之间的分子运动加快,反相微乳的胶束破坏变大,蛋白质的后萃率增加。当温度继续升高,萃取率反而开始下降,且过高的温度会导致蛋白变性[25]。因此,三种反相微乳体系的最佳萃取温度为40 ℃。
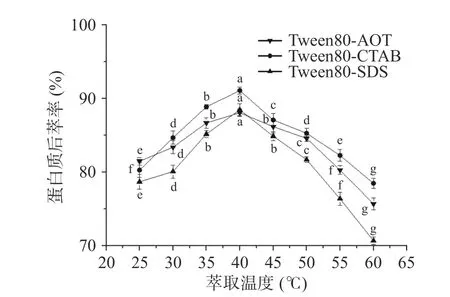
图9 萃取温度对后萃率的影响Fig.9 Effect of temperature on the backward extraction
2.2.4 后萃正交试验 从表7 中正交试验结果可以看出,影响Tween80-AOT、Tween80-CTAB 和Tween80-SDS 三种反相微乳体系后萃率的主次顺序分别是萃取温度>KCl 浓度>缓冲液pH、KCl 浓度>缓冲液pH>萃取温度和萃取温度>KCl 浓度>缓冲液pH。且从表8~表10 中可以看出,三个因素对三种反相微乳体系都有显著影响。实验结果表明,在KCl 浓度为1.2 mol/L、pH 为11.0、萃取温度为50 ℃的条件下,茶渣蛋白在Tween80-AOT 微乳体系中的后萃率最高(91.53%),则三个因素的最佳组合是:A3B2D3。Tween80-CTAB 微乳体系中的三个因素的最佳组合是:A3C3D2,即KCl 浓度为1.2 mol/L,pH 为7.0,萃取温度为40 ℃,达到最高后萃率94.78%。表中观察到Tween80-SDS 微乳体系中茶渣蛋白的最佳后萃取条件为KCl 浓度1.2 mol/L,pH11.0,提取温度30 ℃,则三个因素的最佳组合是:A3B2D1,在最佳条件下茶渣蛋白的后萃率为87.72%。
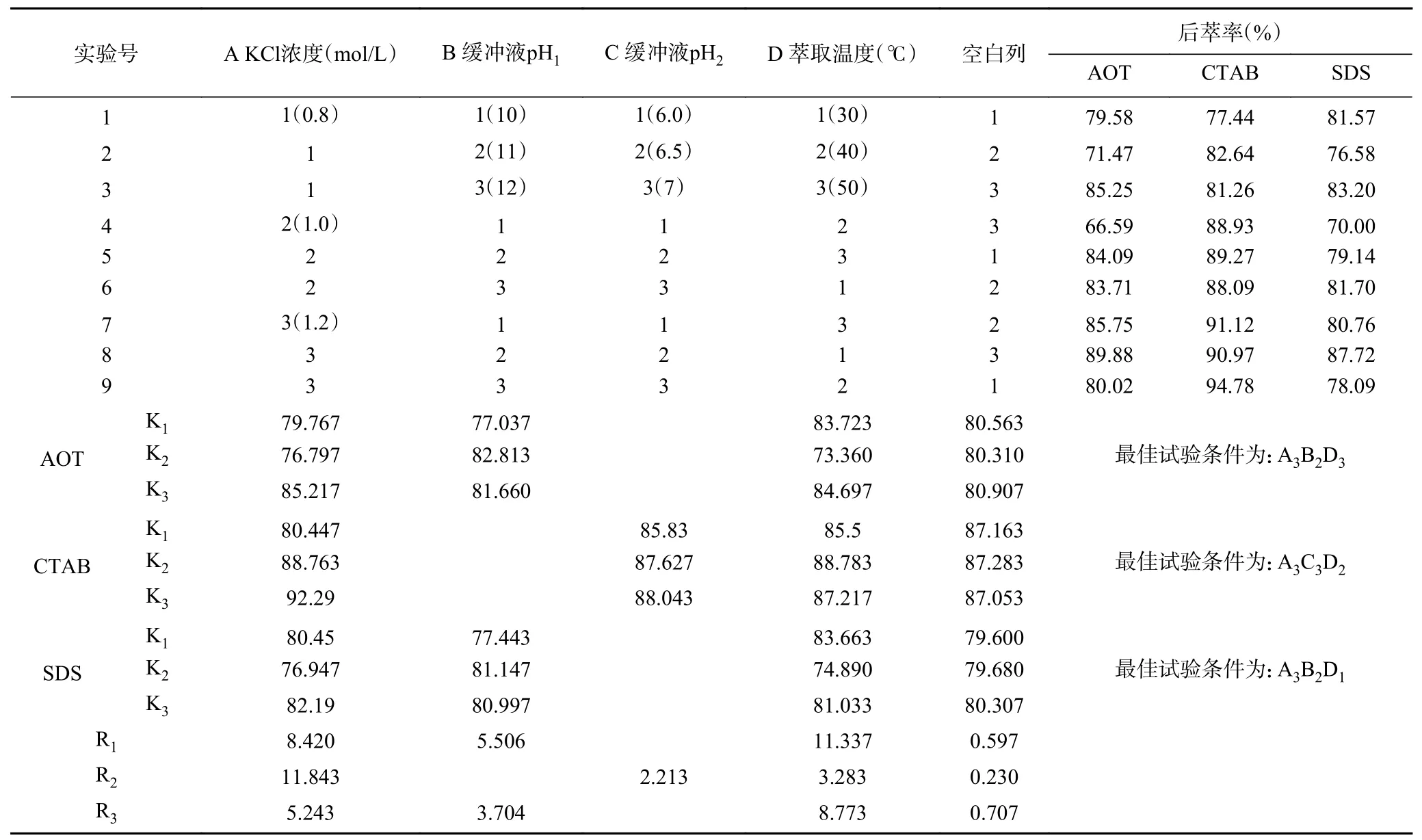
表7 后萃正交试验安排与结果Table 7 Arrangement and results of backward extraction orthogonal experiment
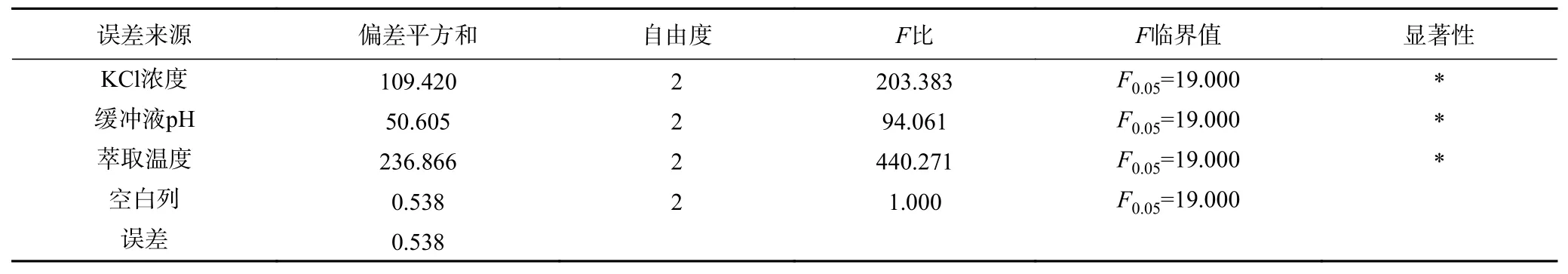
表8 Tween80-AOT 体系后萃正交试验方差分析结果Table 8 Significant analysis on backward extraction orthogonal test of Tween80-AOT system
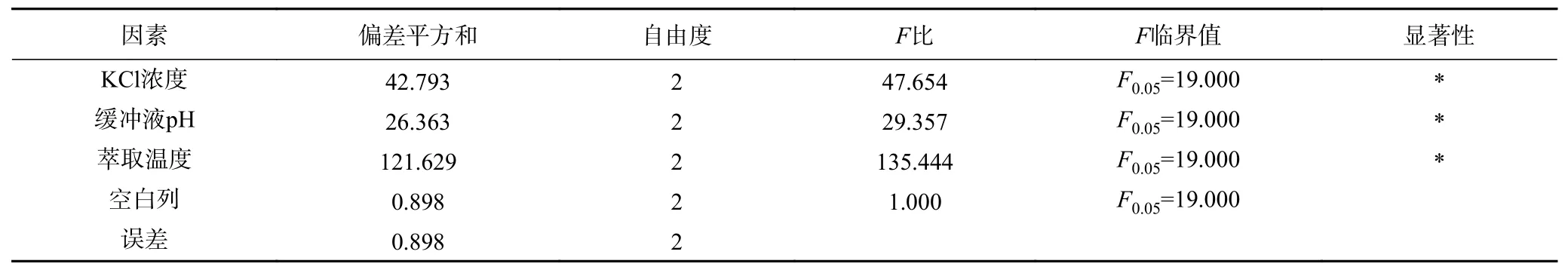
表10 Tween80-SDS 体系后萃正交试验方差分析结果Table 10 Significant analysis on backward extraction orthogonal test of Tween80-SDS system
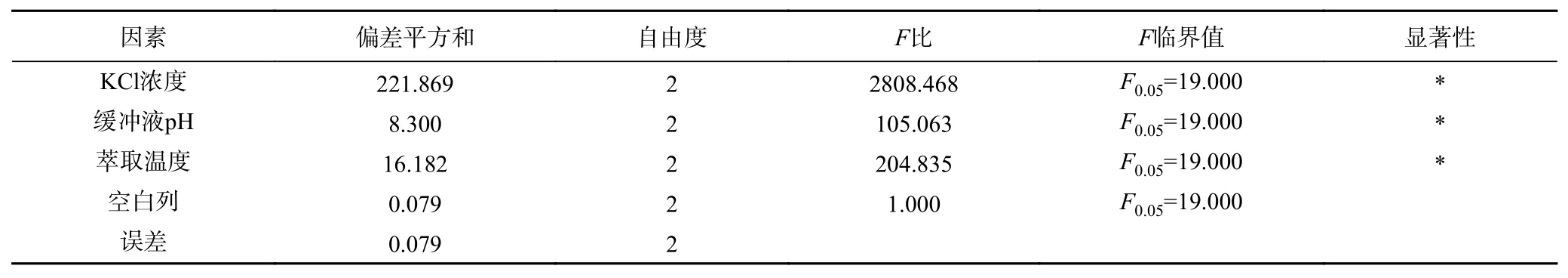
表9 Tween80-CTAB 体系后萃正交试验方差分析结果Table 9 Significant analysis on backward extraction orthogonal test of Tween80-CTAB system
2.3 茶蛋白分子量测定结果
图10展示了用Tween80-CTAB 反相微乳提取茶渣蛋白的SDS-PAGE 电泳图。在茶渣蛋白的光谱中,有两条带子,两条带子的分子量分别为27.18 和20.85 kDa,小于碱法提取茶渣蛋白的分子量。在以前的工作中,我们研究了不同方法(反相微乳法、碱法和酶法)提取的茶渣蛋白的特性,结论表明,反相微乳法提取的茶叶蛋白与碱法和酶法提取的蛋白相比,具有更好的溶解性和乳化性等功能特性[23]。根据SDS-PAGE 电泳的结果可知,碱溶法提取的茶渣蛋白的分子带多于反转微乳法提取的茶叶蛋白。因此,可以知道反相微乳可以选择性地提取高活性的茶渣蛋白。
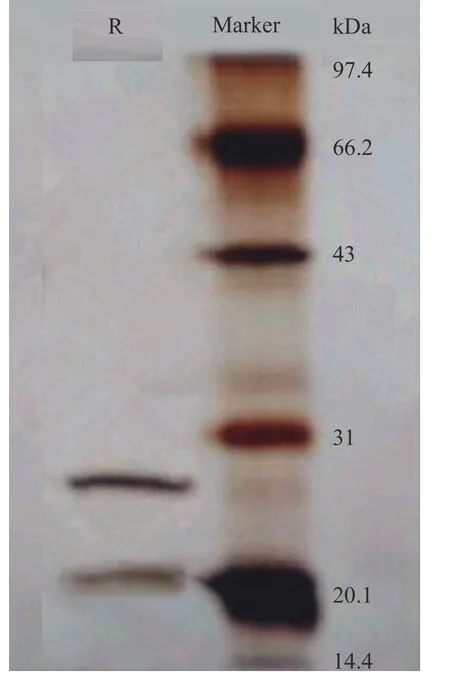
图10 茶渣蛋白分子量Fig.10 Molecular weight of tea residues protein
3 结论
本研究用非离子型表面活性剂(Tween80)和离子型表面活性剂(AOT、SDS、CTAB)制备了3 种混合表面活性剂的反相微乳体系用于提取茶渣蛋白。实验结果表明,从茶叶残渣中提取高活性的茶叶蛋白,Tween80-CTAB 反相微乳是最佳选择,其最佳提取条件为表面活性剂浓度0.10 mol/L,离子型表面活性剂含量70%,pH13.0,W025,萃取温度40 ℃,萃取时间40 min。在这种条件下,茶渣蛋白的前萃率为16.17%。然后在最佳条件(氯化钾浓度为1.2 mol/L,pH7.0,提取温度40 ℃)下进行后萃实验后,得到其最佳后萃率为94.78%。这些数据证明了用反相微乳法从茶叶残渣中提取蛋白质的可行性,为如今大量存在的废弃茶渣提供了一个切实可行的处理方法,同时也减少了废弃茶渣对环境的污染。另一方面,未来的研究还需要进一步提高茶渣蛋白的前萃率,使茶渣能够得到更充分的利用。SDS-PAGE 结果显示,反相微乳可以选择性地提取分子量较小的高活性茶渣蛋白,这可能是前萃率较低的原因。
