细胞代谢调控网络
2022-05-20赵文涛欧阳聪王慧慧张凤琼赵国辉曹婷艳李勤喜
江 彬,赵文涛,欧阳聪,王慧慧,张凤琼,赵国辉,曹婷艳,李勤喜
(厦门大学生命科学学院,细胞应激生物学国家重点实验室,福建 厦门 361102)
细胞是生命活动的基本单位,必须要进行一系列生物学活动才能发挥正常的功能,如生长、分裂、运动、分泌、细胞间交流等,而这些活动的基础就是物质和能量的交换.细胞是通过化学反应来获取物质和能量的,这些化学反应构成了很多代谢通路,不同的通路之间通过交叉对话构成了复杂的细胞代谢网络(物质和能量代谢)[1].
为了方便理解和学习,可将组成代谢网络的代谢通路分为合成代谢和分解代谢.两者是密不可分的,很多代谢通路随着细胞生理状态的变化有时担任合成代谢的任务,有时又扮演分解代谢的角色,这类代谢通路称为两用代谢通路,如糖酵解和三羧酸(tricarboxylic acid,TCA)循环.不同器官在合成和分解代谢的调控上密切配合,如当机体处于长期饥饿状态且肝糖原消耗殆尽的情况下,肝脏会通过糖异生途径(糖酵解的逆反应)生成葡萄糖,分泌到血液中供大脑等肝外组织利用,而这些组织则可以通过糖酵解为其提供能量.虽然细胞代谢网络异常复杂,但是仍有规律可循.对高等需氧生物而言,其能量提供主要是通过三大营养物质(糖、脂类和蛋白质)的分解代谢而实现的.糖酵解、TCA循环、氧化磷酸化等重要的代谢通路称为核心代谢通路.
为了适应不断变化的外部环境,机体必须随时相应地调节细胞的生物学活动,而机体对细胞生物学活动的调节则基于其对细胞代谢网络的调控.通过调节使细胞的能量状态在正常生理情况下始终处于一定范围内的动态平衡,即为细胞能量代谢的稳态.需要说明的是,能量代谢始终伴随着物质代谢,二者不可孤立,所谓的“能量代谢”只是从能量的角度考察代谢及其调控.
细胞代谢稳态的维持有赖于机体和细胞之间及细胞内部多层次、多方位的立体调节网络,即细胞代谢信号网络.该网络非常复杂,包括内分泌系统(如胰岛素、胰高血糖素、肾上腺素、脂肪因子、胃肠激素、甲状腺素等)[2-3]、各种生长因子信号通路(如胰岛素、表皮生长因子等)[2-3]、抑癌因子(如p53)[4]、致癌因子(如雷帕霉素机制性靶蛋白(mechanistic target of rapamycin,mTOR)、缺氧诱导因子1α(hypoxia-inducible factor-1α,HIF-1α)、细胞源性的骨髓细胞瘤病毒癌基因(cellular-myelocytomatosis viral oncogene, c-MYC)等)[5]及细胞能量水平调节的核心分子单磷酸腺苷激活的蛋白激酶(adenosine monophasphate-activated protein kinase, AMPK)等[1].细胞内各代谢通路之间既分工明确、各司其职,又相互依赖、相互制约、协调统一,这样才能维持细胞代谢的稳态.
1 激素对机体代谢的调节
神经内分泌系统起着调节各器官的功能、合理分配能量供应的作用.内分泌系统由内分泌腺组成,内分泌腺包括下丘脑、垂体、胰腺、肾上腺等.内分泌腺分泌激素,激素进入血液被运送到靶器官,在靶器官的细胞表面或细胞核内与其特异性受体结合后启动细胞内的信号转导系统,从而调节细胞的能量代谢及生命活动.肝脏是机体能量代谢的核心器官(三大营养物质都在肝脏进行代谢转化),在能源物质的储存、处理、加工及能量分配中起重要作用.大脑是机体的一个重要的耗能器官,神经元主要以葡萄糖或β-羟基丁酸(酮体的组成之一)为燃料.近年来有研究表明大脑也可以利用乳酸作为燃料[6].由于肝脏、骨骼肌及脂肪组织在能量的储存、分配及利用中起关键作用,三大营养物质在这些组织中的代谢受内分泌系统的严格调控[2].调控代谢的激素很多,因篇幅限制,本文只讨论胰岛素、胰高血糖素和肾上腺素对代谢的调节作用.
1.1 胰岛素
在正常生理状态下,人体血糖维持在60~90 mg/mL(3.33~5 mmol/L)才能保证各器官的正常功能,血糖过高或过低都会导致严重的疾病,甚至死亡.胰岛素和胰高血糖素在维持血糖的稳态中起重要作用.胰岛素是由胰岛β细胞分泌的一种激素,其主要作用是在机体进食后促进糖的储存和利用,从而降低血糖,维持血糖的稳态.进食后胰岛β细胞内三磷酸腺苷(adenosine triphosphate,ATP)水平升高,使K+外流,细胞去极化,进而引起Ca2+内流,导致胰岛素的分泌[7].此外,进食后从肠道分泌的肠促胰岛素可刺激胰岛素分泌,有研究表明其引起的胰岛素分泌能力约占全部胰岛素分泌量的50%~70%[8].葡萄糖依赖性促胰岛素多肽(glucose-dependent insulinotropic polypeptide,GIP)和胰高血糖素样肽-1(glucagon-like peptide 1,GLP-1)是摄入各种营养素后从肠道分泌的两种主要肠促胰岛素激素,以葡萄糖浓度依赖的方式刺激胰腺β细胞分泌胰岛素[9].GIP和GLP-1被二肽基肽酶-4(dipeptidyl peptidase-4,DPP-4)降解,并迅速失去其生物活性[10].GIP和GLP-1的作用分别由它们的特异性受体GIPR和GLP-1R介导,这两种受体在胰腺β细胞以及各种组织和器官中表达.目前,靶向肠促胰岛素受体的药物已取得重大进展,如GLP-1R激动剂可稳定激活GLP-1R信号[11],起到很好的降糖作用.另外,DPP-4的抑制剂通过延缓GIP和GLP-1的降解,增强其促胰岛素分泌的作用,亦取得了良好的临床表现[10].
胰岛素与肝脏细胞、肌肉细胞和脂肪细胞表面的胰岛素受体(insulin receptor,IR)结合激活IR底物1(IR substrate 1,IRS-1),被激活的IRS-1与p85调节亚基及磷脂酰肌醇3-激酶(phosphotidylinositol 3 kinase,PI3K)结合并激活PI3K,被激活的PI3K将磷脂酰肌醇(4,5)二磷酸(phosphatidylinositol(4,5)-biphosphate,PIP2)转变为磷脂酰肌醇(3,4,5)-三磷酸(phosphati-dylinositol(3,4,5)-trisphosphate,PIP3),PIP3通过丙酮酸脱氢酶激酶1(pyruvate dehydrogenase kinase 1,PDK1)激活蛋白激酶B(protein kinase B,又称为AKT),进而引起一系列使血糖降低的生理效应(图1):1)通过使GLUT4从细胞浆转移到细胞膜而激活该转运蛋白的功能,从而加速血液中的葡萄糖向肌肉和脂肪组织的转运[12];2)上调肝脏组织中GCK的表达水平,促进糖酵解[13];3)激活的AKT进而磷酸化糖原合酶激酶3β(glycogen synthase kinase 3β,GSK-3β)的Ser9位点并抑制其激酶活性,解除其对肝脏和肌肉组织中GS的抑制,促进糖原合成[13];4)抑制GPA的活性,从而抑制糖原降解[13];5)激活肝脏细胞和肌肉细胞中的PFK2,从而促进糖酵解,同时激活丙酮酸脱氢酶复合体(pyruvate dehydrogenase complex,PDC)的活性,使丙酮酸迅速转化成acetyl-CoA,为脂肪酸的合成提供原料;6)在肝脏细胞中激活ACC,促进脂肪酸的合成[14];7)在脂肪组织中激活脂蛋白酯酶,促进脂蛋白如VLDL运输的脂类进入脂肪组织,并及时合成TAG加以储存[15].总之,胰岛素的主要功能是使葡萄糖以糖原和脂肪的形式储存起来,降低血糖使其维持在正常水平.

GLUT.葡萄糖转运蛋白;GCK.葡萄糖激酶;GPA.糖原磷酸化酶A;acetyl-CoA.乙酰辅酶A;ACC.acetyl-CoA羧化酶;GS.糖原合酶;G6P.葡萄糖-6-磷酸;F6P.果糖-6-磷酸;PFK2.磷酸果糖激酶2;VLDL.极低密度脂蛋白;TAG.甘油三酯.
1.2 胰高血糖素
胰高血糖素是一种由29个氨基酸组成的肽素,具有多种生物学功能,如维持葡萄糖稳态[16].胰高血糖素的分泌响应于多种代谢信号的变化[17-18],如血糖浓度变化[19]、特定氨基酸浓度变化[20]、脂肪酸浓度以及压力应激变化(如交感神经系统的激活)[21].GCK是葡萄糖浓度的感受器,它可以敏锐感知葡萄糖浓度变化,及时调控胰岛素和胰高血糖素的分泌,维持血糖稳定[22].在低血糖情况下,GCK活性迅速下降,启动胰岛α细胞的胰高血糖素释放机制,使胰高血糖素分泌增加,通过与肝脏上的七重跨膜G蛋白偶联受体结合发挥功能[23],可通过抑制糖原合成和糖酵解以及促进糖原分解和糖异生来增加肝脏中葡萄糖的产生[24-25].在长期饥饿的情况下,血液中的葡萄糖不能通过进食加以补充,但外周组织仍然要利用血液中的葡萄糖作为燃料,因此,随着饥饿的持续,血糖水平会逐渐降低.在正常情况下血糖是大脑的主要燃料,血糖太低时大脑的功能会因缺乏能量而出现异常,因此机体要维持正常的生理功能就必需维持血糖的稳态.在饥饿的情况下,机体分泌胰高血糖素,胰高血糖素与细胞质膜上的跨膜受体结合[26],导致受体构象变化,从而激活三聚体三磷酸鸟苷结合调节蛋白(trimeric guanosine triphosphate-binding regulated protein,G蛋白)偶联蛋白,进一步导致腺苷酸环化酶(adenylyl cyclase,AC)被激活,环状单磷酸腺苷(cyclic adenosine monophosphate,cAMP)水平升高,进而激活蛋白激酶 A(protein kinase A,PKA)和 cAMP 反应元件结合蛋白(cAMP response element binding protein,CREB)[25].PKA 和CREB引起以下一系列适应性代谢改变,以满足机体对能量的需求,如使血糖升高、脂肪动员等(图2):1)PKA磷酸化并抑制PK[27]和PFK1[24],进而抑制肝脏细胞的糖酵解;2)PKA磷酸化并抑制肝脏细胞中GS的活性,阻止糖原合成[13];3)PKA 磷酸化磷酸化酶激酶从而激活肝脏细胞中GPA的活性,启动糖原分解级联反应[28],促进肝糖原转化为游离的葡萄糖并释放入血液[13];4)CREB诱导PEPCK的转录,从而激活PEPCK[16],同时PKA 磷酸化并激活FBPase-2[24],进而促进肝细胞的糖异生;5)激活脂肪细胞中的PKA,PKA进一步磷酸化并激活HSL加速脂肪酸的动员[29];6)抑制肝细胞中的ACC,促进脂肪酸转变为酮体,为脑等肝外组织提供能量[14,29].

WAT.白色脂肪组织;PK.丙酮酸激酶;F-1,6-P.果糖-1,6-二磷酸;PEP.磷酸烯醇式丙酮酸;PEPCK.PEP羧化激酶;FBPase-2.果糖-2,6-二磷酸酶;F-2,6-P.果糖-2,6-二磷酸;Pi.磷酸根;HSL.激素敏感性酯酶.
1.3 肾上腺素
在应激条件下机体会发生一系列生理变化,如心跳加速、血压上升、呼吸加速、脂肪动员加快、肌肉组织中糖的分解加速等应激反应.肾上腺素是由髓质分泌引起应激反应的最主要的内分泌激素,受内脏神经直接支配,当大脑皮层接收到刺激信号,肾上腺素分泌量增加,经血液循环运送到其他组织,和靶细胞表面的肾上腺素受体(G蛋白偶联型,主要包括肾上腺素α受体和β受体)结合,促使受体结构发生改变,将受体转化为激活形式[30],被激活的肾上腺素受体与G蛋白结合,G蛋白中的二磷酸鸟苷(guanosine diphosphate,GDP)被三磷酸鸟苷(guanosine triphosphate,GTP)取代而激活,被激活的G蛋白从受体中释放出来激活腺苷酸环化酶, 腺苷酸环化酶被激活后,将大量ATP分子转化为第二信使cAMP.在肝脏细胞或骨骼肌细胞中cAMP可以结合并激活PKA,活化的PKA进一步将GPB转化为其活性形式GPA,进而触发糖原的分解,为肌肉细胞提供G6P,为肝细胞提供G6P和游离葡萄糖;此外,活化的PKA也能磷酸化GS,将其从活性形式转化为非活性形式,从而抑制糖原合成[31].在脂肪细胞中,肾上腺素刺激cAMP生成,使PKA磷酸化,进而激活HSL,促进脂肪动员,加速脂肪分解,脂肪分解的产物甘油可经糖异生合成葡萄糖.在心肌细胞中,肾上腺素通过cAMP激活PKA,后者能磷酸化胞膜L-型钙离子通道,增加收缩期心肌细胞的Ca2+内流和肌浆网的Ca2+释放,增强心肌收缩能力,缩短收缩间隔,导致心率和血压升高,增强为组织运输氧气和营养物质的能力[32].为了防止肾上腺素受体信号的持续过度激活,机体可利用β-arrestins衔接蛋白抑制肾上腺素受体对下游信号通路的激活,其原理为:被激活的β-肾上腺素受体能被β-肾上腺素受体激酶磷酸化,β-arrestins 能识别磷酸化的β-肾上腺素受体并与之结合,通过空间位阻效应抑制G蛋白与β-肾上腺素受体结合[33],从而终止或减弱β-肾上腺素介导的信号通路.
2 细胞代谢中的重要信号通路
2.1 mTOR信号通路与合成代谢
rapamycin是近半个世纪以前从吸水链霉菌(Streptomyceshygroscopicus)中分离得到的强效抗真菌和抗免疫细胞增殖的药物,因具有很强的抗增殖性使其成为研究细胞增殖调控的常用工具.在20世纪90年代初通过酵母菌(Saccharomyces)的基因测序发现了rapamycin 的两个靶基因TOR1和TOR2[34-35],其突变株可以逃离细胞周期捕捉.随后在哺乳动物中也发现了TOR基因,即mTOR[36].mTOR 是高度保守的丝氨酸/苏氨酸蛋白激酶(serine/threonine protein kinase,STK),属于磷脂酰肌醇激酶相关激酶(phosphatidylinositol kinase-related kinase,PIKK)家族.mTOR 参与组成 mTORC1和mTORC2这两种多蛋白复合物[37],但rapamycin只抑制mTORC1中mTOR的活性却不能抑制mTORC2中mTOR的活性.为避免理解混乱,目前已把mTOR以前的命名“雷帕霉素的哺乳动物靶蛋白”(mammalian target of rapamycin,mTOR)改为“雷帕霉素机制性靶蛋白”(mechanistic target of rapamycin,mTOR),英文缩写保持不变.mTORC1 在细胞的增殖和分化以及能量代谢中均起关键的调节作用.生长因子如胰岛素与其受体结合后激活受体酪氨酸酶的活性,通过PI3K-AKT途径或Ras-Raf-Erk途径激活mTORC1[38].mTORC1被激活后能激活真核细胞翻译起始因子4E(eukaryotic translation initiation factor 4E,eIF4E)[39-40]、核糖体S6蛋白激酶1(ribosomal protein S6 kinase beta-1,S6K1)[41]和固醇调节元件结合蛋白1(sterol regulatory element-binding protein 1,SREBP1)[42].eIF4E和S6K1被激活后可以促进核糖体的生物合成,激活cyclin D1、c-MYC、周期蛋白依赖性激酶2(cyclindependent kinase 2,CDK2)等与细胞增殖相关蛋白的翻译,刺激基质金属蛋白酶9(matrix metalloproteinase 9,MMP9)的翻译,增强HIF-1α的表达,从而促进糖酵解及血管生成.mTORC1通过这些作用促进蛋白合成、刺激细胞分裂及增殖、增强糖酵解、促进肿瘤血管形成及转移.SREBP1被mTORC1激活后能转录激活参与磷酸戊糖旁路及脂肪合成的一系列酶的转录,促进脂肪的合成及储存[43].
目前关于mTORC1激活条件及详细分子机制方面的研究均取得了很大进展(图3).mTORC1的激活有3个最基本的条件:1)mTORC1主要促进消耗能量的合成代谢,抑制产生能量的分解代谢,因此mTORC1激活的先决条件是机体在整体水平上有足够的能量且分泌较高水平的生长因子;2)细胞本身要有充足的能量供应;3)mTORC1激活后的一大功能是促进蛋白质的合成,而氨基酸是蛋白质合成的原料,因此mTORC1的激活必须有足够浓度的氨基酸,尤其是支链氨基酸.这3个条件必须同时具备,mTORC1才能被激活,可见mTORC1的重要性.生长因子和能量水平调控mTORC1的靶点是TSC1/2.TSC1/2是一种GTP 酶活化蛋白(GTPase-activating protein,GAP),它被激活后能够抑制mTORC1,其机制是TSC1/2的GAP酶活性能使mTORC1的直接激活蛋白Rheb由GTP 偶联的活性形式转化为GDP偶联的失活形式[44].AKT被胰岛素及生长因子激活后能直接磷酸化TSC1/2使其失活,从而激活mTORC1.细胞缺乏能量时AMPK被激活,AMPK可以磷酸化TSC1/2使其激活,从而抑制mTORC1;而在细胞能量供应充足的情况下AMPK失活,其对mTORC1的抑制作用也随之解除.缺氧一方面导致细胞ATP水平降低,激活AMPK、抑制mTORC1;另一方面通过激活REDD1调节蛋白,从而抑制mTORC1.另外,DNA损伤刺激p53表达,后者可以通过激活REDD1和AMPK间接抑制mTORC1.

RTK.受体酪氨酸激酶;Grb2.生长因子受体结合蛋白2;SOS.Sevenless之子蛋白;NF1.神经纤维瘤蛋白1;PTEN.磷酸酶和张力蛋白同系物;Raf.RAF原癌基因STK;ERK.丝裂原活化蛋白;MEK.双特异性ERK激酶;RSK.核糖体蛋白S6激酶;PDK1.3-磷酸肌醇依赖蛋白激酶1;LKB1.丝氨酸/苏氨酸激酶11;FKBP12.FKBP脯氨酰异构酶1A;GβL.mTOR相关蛋白LST8同系物;DEPTOR.含mTOR相互作用蛋白的DEP结构域;4E-BP1.真核翻译起始因子eIF4e结合蛋白1;ATG13.自噬相关蛋白13;ULK1.Unc-51类自噬激活激酶1;Sin1.应激激活蛋白激酶相互作用蛋白1;PRR5.脯氨酸蛋白5;Rictor.雷帕霉素不敏感mTOR伴侣蛋白;Lipin1.磷脂酸磷酸酶1;SGK1.血清和糖皮质激素调节激酶 1;PKCα.蛋白激酶Cα;TSC1/2.结节性硬化症复合物1/2;Rheb.富集于大脑的Ras同系物;REDD1.发育和DNA损伤反应1;Rag.Ras相关的GTP酶;Ragulator.Rag调节因子;v-ATPase.v型三磷酸腺苷酶.
氨基酸激活mTORC1的原理比较复杂,涉及到Rag蛋白[45-46]、Ragulator复合体[47]、v-ATPase[48]及其他具有鸟嘌呤核苷酸交换因子(guanine nucleotide exchange factor,GEF)和GAP活性的调节蛋白[49-50].Ragulator是由LAMTOR 1(late endosomal/lysosomal adaptor,MAPK and mTOR activator 1)、LAMTOR2、LAMTOR3、LAMTOR4和LAMTOR5组成的异源五聚体,能进一步与v-ATPase形成复合体,定位在晚期溶酶体上.Rag蛋白有A、B、C、D 4种亚型,其中A与B相似,C与D相似,在细胞中一般是A/B与C/D组成异源二聚体.v-ATPase与Ragulator形成的复合体能将Rag异源二聚体募集到溶酶体表面.当细胞氨基酸浓度较高时溶酶体中的氨基酸会作用于v-ATPase引起Ragulator的构象变化,激活其针对RagA/B的GEF活性,从而使Rag二聚体中的A/B与GTP结合;同时抑癌因子FLCN(folliculin)针对RagC/D的GAP活性也被激活,使Rag二聚体中的RagC/D与GDP结合.此时,RagA/B与RagC/D形成的二聚体被激活后能与mTORC1中的Raptor结合,从而将mTORC1定位到溶酶体上,进而被已经定位到溶酶体上的mTORC1激活蛋白Rheb激活[51-52].
近年来的研究进一步阐明了mTOR感应氨基酸浓度的机制(图4).Sabatini课题组揭示了氨基酸通过多种方式介导mTORC1活化的机制:首先他们鉴定了一种氨基酸转运体SLC38A9,这是一种定位于溶酶体上的精氨酸转运体,作为溶酶体跨膜蛋白,它能够以一种氨基酸敏感的方式与Rag蛋白及Ragulator相互作用,将mTORC1定位到溶酶体上[53].另外,他们还发现定位于胞浆的GATOR1蛋白具有针对RagA的GAP活性,它被激活后能够抑制mTORC1的溶酶体定位.当细胞内的氨基酸充足时,亮氨酸能够与Sestrin2结合来破坏Sestrin2与GATOR2的相互作用,使得GATOR2处于激活状态,激活的GATOR2进一步解除GATOR1对mTORC1的抑制[54].与亮氨酸不同的是,精氨酸通过以30 μmol/L的解离常数与CASTOR1结合来破坏CASTOR1与GATOR2的相互作用,使得GATOR2处于激活状态,从而激活mTORC1[55].Kim课题组[56]则发现,LARS1能够感知亮氨酸浓度并调节其与RagD的相互作用,行使GAP功能,使RagDGTP转化为活化态RagDGDP,从而激活mTORC1通路.此外,Kim课题组[57]还发现LARS1对mTORC1的调控作用依赖于葡萄糖的存在,具体机制是:葡萄糖存在时,LARS1可以感知到亮氨酸的存在,使亮氨酸与tRNA进行共价结合,参与翻译过程,同时通过RagD激活mTORC1通路;葡萄糖缺乏时,ULK1磷酸化LARS1降低其与亮氨酸的结合能力,从而减弱对翻译过程和mTORC1的调控作用,蛋白质的翻译被抑制进而引发细胞自噬,帮助细胞短暂适应缺糖环境.值得关注的是,赵世民课题组[58]研究表明:氨基酰tRNA合成酶也兼有氨基酰转移酶的活性,当细胞内的某种氨基酸水平升高时,结合了这种氨基酸的tRNA合成酶会与胞内特定的蛋白结合,将氨基酰基转移到蛋白特定的赖氨酸残基上对其进行氨基酰化修饰.如细胞内的亮氨酸含量较高时,可以通过LARS1修饰RagA,使其更容易转变为RagAGTP形式,从而促进mTORC1的激活.这些研究成果为阐明氨基酸激活mTOR信号通路填补了重要一环.

CASTOR.胞质精氨酸传感器;GATOR.Rag二聚体的上游关键调控复合物(能够水解Rag二聚体从而影响mTORC1在溶酶体的定位);LARS.亮氨酸-转运RNA(tRNA)合成酶.
总之,mTORC1主要是在营养物质和能量充足的情况下激活合成代谢,消耗ATP,抑制细胞自噬,促进细胞存活和增殖.因此,mTORC1的过度激活与肿瘤的发生发展密切相关,被认为是一个很重要的抗肿瘤靶点[38].自2007年起,雷帕霉素的两种衍生物被开发用于通过抑制mTORC1来治疗癌症,分别为辉瑞公司的坦罗莫司(temsirolimus)和诺华公司的依维莫司(everolimus).
2.2 AMPK信号通路与分解代谢
AMPK是细胞内的能量感受器,在真核细胞生物中广泛存在.各种导致细胞内单磷酸腺苷(adenosine monophosphate,AMP)/ATP或二磷酸腺苷(adenosine diphosphate,ADP)/ATP比值升高的因素如饥饿、缺氧等均可引起AMPK活化.AMPK活化后抑制消耗ATP的合成代谢过程,启动生成ATP的分解代谢过程,从而维持机体能量代谢的稳态.
AMPK是进化过程中高度保守的STK,最初是作为脂肪合成代谢中的关键酶ACC和胆固醇合成代谢中的关键酶3-羟基-3-甲基戊二酸单酰辅酶A还原酶(3-hydroxy-3-methyl glutaryl-CoA reductase,HMGR)的上游调节因子被发现的[59].AMPK是由α、β和γ亚基组成的异源三聚体:其中α亚基具有催化活性;β亚基连接α和γ亚基,是形成稳定有活性的三聚体复合物所必需的;γ亚基的Bateman结构域包含AMP的结合位点.AMPK的活性受变构剂和磷酸化的双重调节.如图5所示,当细胞处于饥饿状态时AMP的浓度升高,和AMPK的γ亚基结合,通过构象的改变引起α亚基的Thr172位点暴露,此时AMPK的上游激酶LKB1、CaMKK和TAK1等能使Thr172位点磷酸化,从而激活AMPK的激酶活性[60-61].
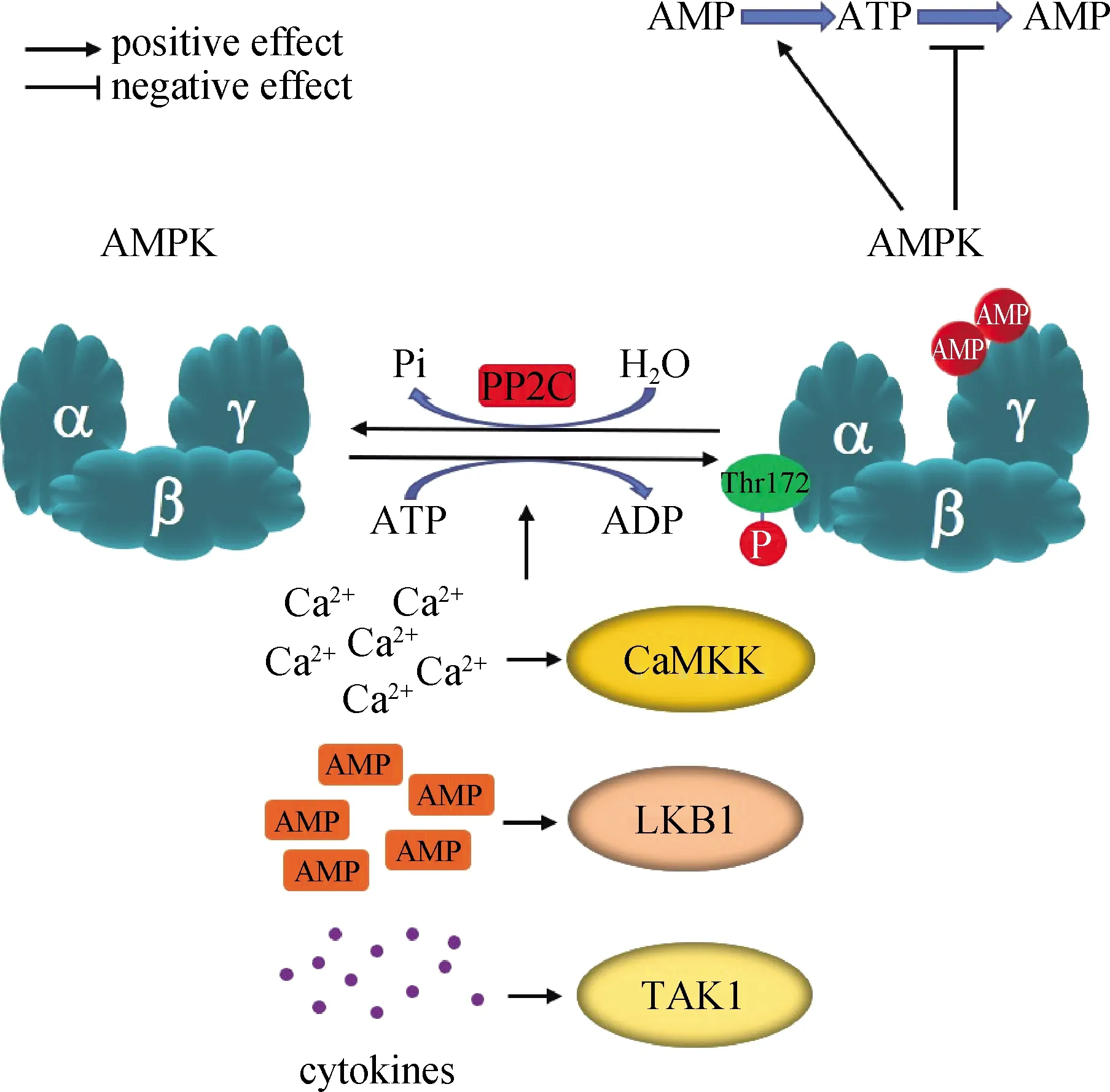
CaMKK.Ca2+/钙调蛋白依赖性蛋白激酶激酶;LKB1.肝脏激酶 B1;PP2C.蛋白磷酸酶2C;TAK1.转化生长因子β相关激酶1.
近年来的研究表明AMPK被激活后可以在整体水平和细胞水平两个层面调控能量代谢的稳态[62].在整体水平上,AMPK主要通过下丘脑调控能量平衡[62],如图6所示:下丘脑弓状核中AMPK的激活可以刺激食欲引起进食活动,下丘脑腹内侧核中AMPK的激活可以抑制交感神经系统,从而抑制交感神经兴奋引起的能量消耗.在饱食的情况下,一方面脂肪细胞会分泌瘦素,抑制弓状核中AMPK的激活,从而抑制进食活动,防止机体摄入过多的能源物质;另一方面,小肠会分泌GLP-1,甲状腺会分泌甲状腺素,二者均可抑制下丘脑腹内侧核中AMPK的活性,解除其对交感神经的抑制作用,从而增强机体的基础代谢效率,加速能量的消耗.在饥饿的情况下,机体缺乏能量,一方面脂肪细胞会分泌脂联素,另一方面胃会分泌饥饿素,二者均能激活弓状核中AMPK的活性,从而刺激进食活动,为机体补充能量.

T3.3,3’,5-三碘甲状腺原氨酸.
在细胞水平上,AMPK响应细胞能量状态的变化,通过多器官多靶点抑制耗能的合成代谢,促进释放能量的分解代谢,从而维持细胞的能量稳态[63-64],如图7所示.在分解代谢方面AMPK有以下作用:1)上调肌肉细胞GLUT1和GLUT4的表达,促进糖的摄入;2)激活PFK2,促进糖酵解;3)抑制ACC2,激活脂肪酸的氧化分解;4)激活ULK1/2, 启动线粒体的自噬;5)激活脂肪酸转运蛋白CD36,促进脂肪酸的摄入.在合成代谢方面AMPK有以下作用:1)抑制HDAC和CRTC2的活性,从而抑制糖异生途径的关键酶PEPCK和葡萄糖6磷酸酶的表达,最终抑制糖异生;2)使GS失活,从而抑制糖原的合成;3)抑制ACC1的活性,从而抑制脂肪酸的合成;4)转录因子SREBP1C能转录激活脂肪合成的一系列酶,AMPK能磷酸化SREBP1C使其失活,从而抑制脂肪的合成;5)抑制甘油磷酸转乙酰酶,从而阻止三酯酰甘油的合成;6)通过抑制HMGR以抑制胆固醇的合成;7)通过抑制Raptor和激活TSC2以抑制mTOR,从而抑制蛋白质的合成;8)通过抑制转录因子TIFIA以阻止核糖体RNA的合成;9)PGC1α是转录因子PPAR-ϒ 的共激活子,能转录激活线粒体中与能量代谢相关的基因的表达,AMPK可以磷酸化并激活PGC1α,从而增加线粒体的生物合成,促进有氧代谢,增加能量供应.
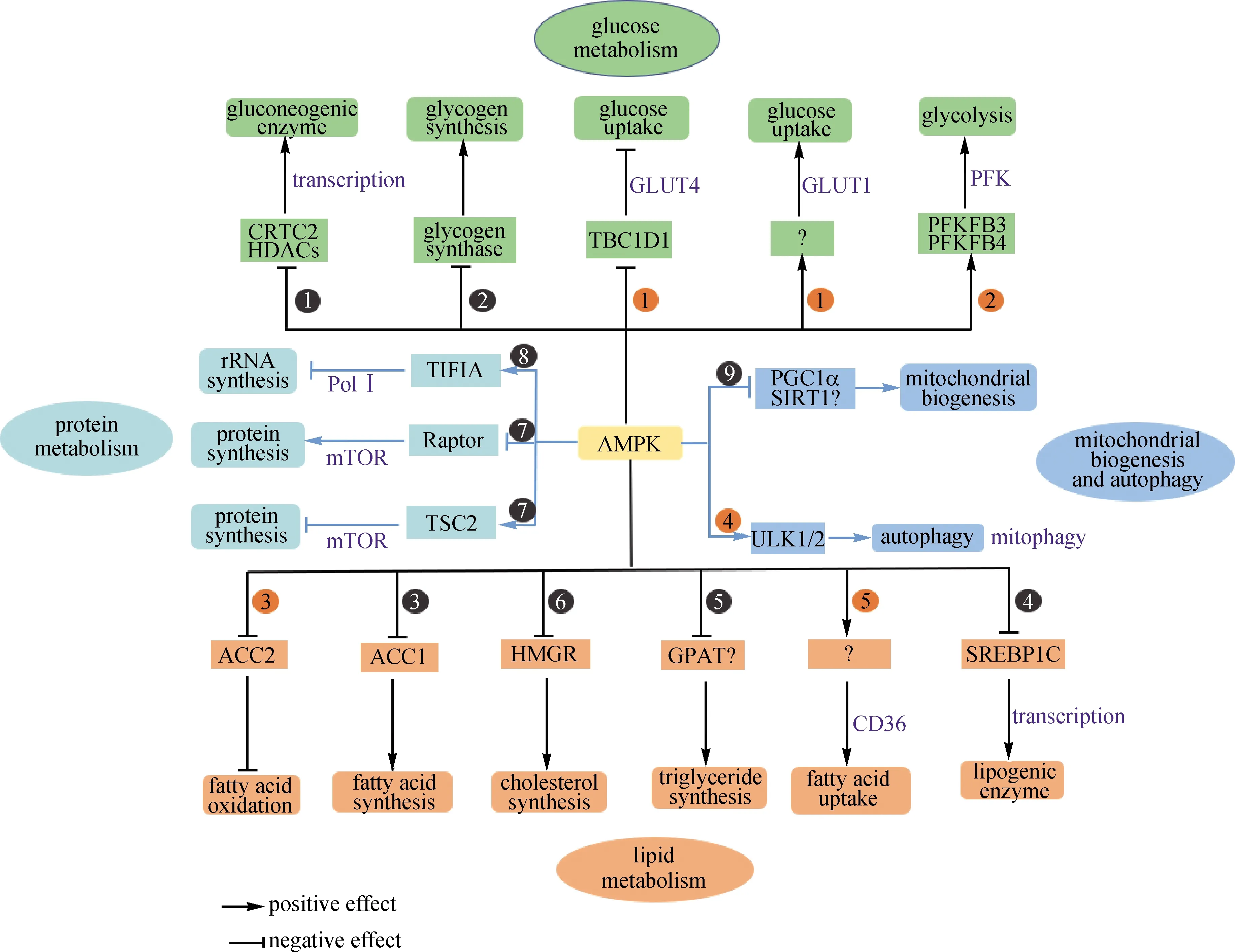
CRTC2.Creb调控的转录共激活因子2;HDAC.组蛋白脱乙酰化酶;TBC1D1.TBC1域家族成员1;PFKFB3.果糖-2,6-二磷酸酶3;TIFIA.RNA聚合酶Ⅰ特异性转录起始因子RRN3;Raptor.mTOR的调节相关蛋白;PolⅠ.RNA聚合酶Ⅰ;PGC1α.过氧化物酶体增殖物激活受体γ(PPAR-γ)共激活因子1α亚基;SIRT1.氧化型辅酶Ⅰ依赖性蛋白脱乙酰酶sirtuin-1;GPAT.3-磷酸甘油酰基转移酶.
鉴于AMPK信号通路在代谢调控中的重要作用,AMPK的活性在细胞内被严格调控.经典理论认为AMPK的活化主要是通过其γ亚基感知细胞内的AMP浓度,通过构象改变以及Thr172位点磷酸化修饰来调节.近几年厦门大学生命科学学院林圣彩课题组的研究结果揭示了细胞在不同能量状态下选择性激活AMPK和mTOR的分子机制(图8).首先他们发现,AXIN作为架构蛋白,在AMP信号的作用下,与AMPK及其上游激酶LKB1三者形成一个复合体,促进LKB1对AMPK的磷酸化激活;且AXIN对于AMPK定位到溶酶体上具有重要的作用,该研究为阐明溶酶体在调控线粒体氧化磷酸化中的作用提供了线索[65].另外,他们研究发现在细胞缺乏能量的情况下,依赖于LKB1的AMPK的激活发生在晚期溶酶体上,其机制是v-ATPase-Ragulator可以将AXIN/LKB1募集到溶酶体表面,从而激活AMPK.由于v-ATPase-Ragulator复合体也是在能量充足的情况下细胞激活mTOR所必须的条件,所以,该复合体成为细胞响应能量状态在合成代谢和分解代谢之间转换的开关[66].接着,他们阐明了在生理状态下机体感受葡萄糖水平的机制.他们发现,葡萄糖水平下降会引起糖酵解的中间产物F-1,6-P水平的下降,该过程进一步地被糖酵解通路上的代谢酶——醛缩酶感应,未结合F-1,6-P的醛缩酶促进v-ATPase、Ragulator和AMPK-AXIN-LKB1之间的动态组合,形成超级复合体,并激活AMPK[67].进一步的机制研究表明,未结合F-1,6-P的醛缩酶可结合阳离子通道蛋白TRPV并抑制其Ca2+的通道活性;Ca2+浓度下降促使醛缩酶-v-ATPase复合体变构,从而抑制v-ATPase,最终启动溶酶体上的AMPK激活途径,引起溶酶体上AMPK的活化[68].他们还发现不同程度的能量缺乏可逐级激活胞内不同区域的AMPK:当细胞处于轻度饥饿时(AMP/ATP比值还没有大的变化),溶酶体上的AMPK最先被磷酸化并激活;当细胞处于中度饥饿时,胞质中的AMPK开始被激活;而当细胞中能量严重匮乏时,线粒体上的AMPK被最终激活.该研究工作首次揭示了不同时空下AMPK激活的现象,对人们更深入、多层次了解AMPK的激活状态具有重要意义[69].

AXIN.轴抑制蛋白1;TRPV.瞬时受体电位阳离子通道亚家族V;ER.内质网.
2.3 AMPK 与mTOR信号通路协同维持细胞能量稳态
AMPK和mTOR分别激活分解代谢和合成代谢,二者只有密切配合、彼此调控才能更好地维持细胞的能量稳态.在机体能量充足的情况下,mTOR通过多条途径激活合成代谢、抑制分解代谢:首先,脂肪细胞会分泌瘦素,瘦素作为内分泌激素在下丘脑控制摄食的中枢中能激活mTOR,进而激活S6K1,S6K1磷酸化弓状核中的AMPK使其失活,从而抑制进食活动;其次,机体会分泌胰岛素等生长因子,通过AKT或ERK通路激活各组织器官细胞中的mTOR,进而直接或间接激活eIF4E、S6K1、SREBP1、cyclin D1、c-MYC、CDK、HIF-1α等,促进生物合成、细胞增殖;同时S6K1能磷酸化AMPK的Ser491位点,抑制Thr172位点的磷酸化,从而通过抑制AMPK的激活,抑制分解代谢[62,70].在饥饿、缺氧、DNA损伤等情况下,AMPK被激活,调节一系列下游靶蛋白激活分解代谢;同时,AMPK可以通过磷酸化激活TSC1/2以及磷酸化失活Raptor而抑制mTOR的激活,从而抑制合成代谢[43].值得一提的是,v-ATPase-Ragulator复合体在不同能量情况下分别激活AMPK和mTOR[66],在细胞处于饥饿情况下,AXIN转运至线粒体促进mTORC1的解离[65-66].因此,该复合体成为细胞响应能量状态在合成代谢和分解代谢之间转换的开关.
3 抑癌因子与致癌因子对能量代谢的调控
在正常生理状态下机体正常细胞主要靠氧化磷酸化提供能量,只有在短暂缺氧的情况下部分组织(如肌肉)可以通过无氧糖酵解提供能量.然而这种正常代谢模式在肿瘤细胞中发生了重大改变.肿瘤细胞对代谢模式进行了重编程,使有氧代谢被抑制而无氧代谢被加强.即使在有氧的情况下,肿瘤细胞仍然靠糖酵解提供能量,这一现象被称为“Warburg效应”,是由德国诺贝尔奖获得者Otto Heinrich Warburg于20世纪30年代提出来的[71].经过近一个世纪的发展,人们揭示了Warburg效应发生的部分分子机制,目前该效应被普遍认为是肿瘤细胞代谢的基本特性之一[71-73].很多致癌基因和抑癌基因编码的蛋白产物参与代谢模式的调控.抑癌因子如p53主要抑制无氧糖酵解,激活TCA循环及氧化磷酸化有氧代谢途径;相反地,致癌因子如HIF-1α和c-MYC等促进无氧糖酵解,而抑制有氧代谢.p53缺失或突变失活及HIF-1α、c-MYC等过度激活都会导致细胞代谢模式从有氧代谢向无氧糖酵解转变,从而促进细胞的恶性转变.
3.1 p53
p53主要通过以下途径抑制糖酵解(图9):1)p53能直接抑制GLUT1和GLUT4的表达[74],也能通过抑制NF-κB间接抑制GLUT3的表达,从而减少细胞对葡萄糖的摄入[75].2)PFK1是糖酵解通路的限速酶之一,该酶能被F-2,6-P变构激活.p53转录激活的一个下游蛋白TIGAR是F-2,6-P的磷酸酶,能使其转化为F6P,因此p53能通过激活TIGAR来抑制糖酵解[76-77].3)PPP能为快速增殖的细胞提供合成核酸所必须的R5P,也能为其他合成代谢途径提供NADPH,肿瘤细胞的PPP非常活跃.p53能抑制PPP的关键酶G6PDH,从而抑制该代谢途径[78].4)p53能抑制MCT1从而抑制乳酸的转运,破坏肿瘤细胞的微环境[79].
p53主要通过以下途径促进有氧代谢(图9):1)PDK1能磷酸化PDC使其失活,而PDC是acetyl-CoA进入TCA循环的关键步骤,p53能通过抑制PDK1而激活PDC[80].2)SCO2能激活电子传递链复合体四(complex Ⅳ)中的关键蛋白细胞色素c氧化酶的合成;AIF也能激活电子传递链,二者均能被p53激活[81-82].3)ME在将苹果酸转化为丙酮酸的过程中产生合成代谢必需的NADPH,p53能通过抑制ME1和ME2进而抑制合成代谢[83].4)p53R2[84]及Mieap在维持线粒体基因组完整性及线粒体质量方面起重要作用[85],p53能通过激活这两种蛋白维持线粒体的正常功能,从而促进有氧代谢[86].5)GLS2可以催化谷氨酰胺水解生成谷氨酸,谷氨酸可进一步通过生成α-酮戊二酸参与TCA循环,p53可以通过转录激活GLS2,从而促进线粒体呼吸和ATP的产生[87-88].
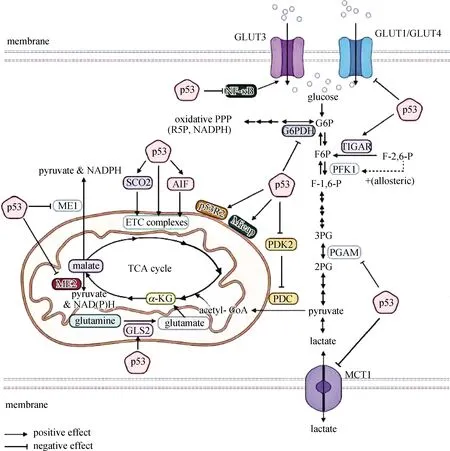
PPP.磷酸戊糖旁路;NADH.还原型辅酶Ⅰ;NADPH.还原型辅酶Ⅱ;p53R2.p53控制的核苷酸还原酶;R5P.核糖-5-磷酸;3PG.3磷酸甘油酸;MCT1.单羧酸转运蛋白1;G6PDH.G6P脱氢酶;ME.苹果酸酶;GLS2.线粒体谷氨酰胺酶2;TIGAR.TP53诱导的糖酵解与凋亡调节蛋白;SCO2.参与细胞色素c氧化酶组装的调节蛋白;AIF.细胞凋亡诱导因子;Mieap.线粒体自噬蛋白;PGAM.新型磷酸甘油酸变位酶;ETC.电子传递链;α-KG.α-酮戊二酸.
另外,需要特别强调的是p53能与AMPK及AKT/TSC/mTOR通路进行交叉对话,从而抑制mTOR,进而抑制合成代谢.首先,p53能通过直接激活AMPK的上游激酶LKB1进而激活AMPK[89],或通过激活Sestrins及AMPK β亚基的转录间接激活AMPK[90-92],AMPK被激活后能抑制mTOR.其次,p53能通过激活AKT的抑制蛋白PTEN而抑制mTOR信号,或通过直接激活TSC1/TSC2复合体抑制mTOR[86].相反地,具有获得性功能(gain of function,GOF)的p53突变体则能抑制AMPK,激活mTOR促进合成代谢和细胞增殖[93].
3.2 HIF-1α 和c-MYC对细胞代谢的调控作用
HIF-1α是细胞在缺氧条件下进行代谢模式转变以适应环境变化的核心分子.在有氧的情况下,HIF-1α被PHD2(prolyl-hydroxylase 2)羟基化,然后被VHL(von Hippel-Lindau)泛素连接酶复合体泛素化,经蛋白酶体途径降解,因此,在正常生理情况下在细胞中几乎检测不到HIF-1α蛋白的表达[94].由于分子氧是PHD2催化羟基化反应的底物之一,所以在缺氧的情况下HIF-1α不能被羟基化,因此不能被VHL泛素化降解,导致HIF-1α在细胞内积累,水平升高[95].而HIF-1α本身是转录因子,其转录激活的下游基因超过100个,如图10所示,这些基因中参与代谢调控的包括:1)GLUT1和GLUT3[96-97];2)参与糖酵解途径的酶,如HK1、HK2、PGK1[98-99]、PKM2[100]和LDHA[101];3)PDK1,能抑制PDC的功能,从而抑制丙酮酸进入TCA循环[102];4)产生NADPH的酶,如IDH1和IDH2[103];5)参与脂肪酸合成的酶,如FASN和Lipin1[104-105];6)Ⅰ型 GS、二磷酸尿苷-葡萄糖焦磷酸化酶(uridine disphosphate-glucose pyrophosphorylase,UGP2)和α-1,4葡聚糖分支酶(1,4-α-glucan branching enzyme,GBE1),促进糖原合成[106];7)脂肪酸结合蛋白3(fatty acid binding protein 3,FABP3)和FABP7,促进脂肪酸的摄入[107].通过激活以上的酶系统HIF-1α促进无氧糖酵解、糖原合成、乳酸产生、脂类摄入及脂类合成,抑制有氧代谢[106,108-110].

HK1/2.己糖激酶1/2;PGK1.磷酸甘油酸激酶1;PKM2.丙酮酸激酶M2;LDHA.乳酸脱氢酶A;PDH.丙酮酸脱氢酶;FASN.脂肪酸合酶;IDH1/2.异柠檬酸脱氢酶1/2.
近几年的研究表明谷氨酰胺代谢在部分肿瘤的生长中起重要作用.在很多肿瘤中HIF-1α过度激活,抑制了PDC,使丙酮酸不能进入TCA循环合成柠檬酸,导致脂肪酸合成受阻,在这种情况下谷氨酰胺便成为脂类合成最主要的原料.谷氨酰胺被GLS转化为谷氨酸后,经TCA循环的逆反应生成柠檬酸,从而进入脂肪酸合成途径[103,113].近年来研究发现PKM2在多数肿瘤中高表达,该酶作为糖酵解通路的最后一个酶,能通过调节无氧代谢和有氧代谢之间的平衡调控合成代谢及分解代谢的稳态,从而促进肿瘤细胞增殖;受体酪氨酸激酶能激活PKM2,PKM2反过来能激活c-MYC及cyclin D1的表达;另外,PKM2也可以作为HIF-1α的共激活因子激活糖酵解通路中一系列酶的转录[114-116].
4 研究细胞代谢网络的意义
细胞能量代谢稳态是细胞其他生物学活动的基础,而细胞能量代谢稳态的维持有赖于细胞能量代谢信号网络的调控.细胞由于基因缺失或突变导致该调控网络失衡,使得细胞能量代谢模式发生改变或能量代谢稳态被破坏,都会引起严重的疾病如癌症、肥胖、糖尿病、心血管疾病等,甚至导致细胞及机体死亡.深入研究细胞能量代谢信号网络的组成成分、各成分的功能及相互调节关系可以为预防及治疗上述疾病提供理论基础及药物靶点,促进基于细胞能量代谢调控的新药的研发,最终为预防及治疗以上由细胞能量稳态被破坏导致的疾病奠定基础.
从代谢的角度看,癌症是一种代谢性疾病,其发生发展的根源是调控代谢的抑癌因子(如LKB1、AMPK、p53、PTEN、TSC等)与原癌基因产物(如AKT、mTOR、HIF-1α、c-MYC等)之间的平衡被打破,引起细胞代谢模式从有氧代谢转变为无氧代谢,细胞合成代谢加速,细胞的适应性增强,这种代谢稳态的变化为细胞恶变及增殖奠定了物质及能量基础.长期以来,人们一直认为细胞能量代谢的改变是伴随癌症的表现,而不是引起细胞癌变的原因.目前研究表明细胞代谢的改变完全有可能直接导致癌症的发生,如IDH1和IDH2的突变就可以导致胶质瘤和白血病等[117].多种人类肿瘤中存在mTOR的过度表达及激活,同时抑癌因子LKB1、PTEN和TSC1/TSC2作为mTOR信号通路中的负向调节因子,在多种肿瘤细胞中也存在缺失表达.LKB1抑癌因子的作用机制就是激活AMPK,进而抑制mTOR信号通路,这使得AMPK 可能成为潜在的抗肿瘤靶点.在许多肿瘤细胞的体外试验中,人们发现AMPK激活剂二甲双胍和5-氨基咪唑-4-甲酰胺核糖核苷酸(5-aminoimidazole-4-carboxamide ribonucleotide,AICAR)有抑制肿瘤细胞增殖的作用[53,118-120].
Ⅱ型糖尿病发生的一个机制是机体出现胰岛素抵抗.当机体营养素水平过剩时, 高浓度的氨基酸或葡萄糖长期刺激使血液中胰岛素浓度增高,进而过度激活mTOR/S6K1通路,S6K1反过来使IRS-1的Ser312、Ser636/639或Ser307位点发生过度磷酸化后抑制胰岛素的信号传导,从而严重干扰insulin/PI3K/AKT的胰岛素信号传导功能,引发机体产生胰岛素抵抗[121-122].在这种情况下胰岛素的分泌不能使GLUT4定位到细胞膜上,血液中的葡萄糖不能被及时吸收入细胞,出现高血糖.二甲双胍和AICAR均能激活AMPK,抑制mTOR/S6K1,从而恢复IRS-1的活性,逆转了细胞的胰岛素抵抗,因此可被用于治疗Ⅱ型糖尿病[123].
肥胖患者的合成代谢远超过分解代谢,主要表现为mTOR过度激活、胰岛素抵抗、血脂异常等多种代谢改变.mTOR过度激活在肥胖发生中起重要作用.mTOR 活化可引起脂质的累积,雷帕霉素能抑制脂肪增长.Um等[122]证实S6K1缺失的小鼠脂类分解作用增强,脂肪组织质量下降;在饮水和进食量无显著性改变的小鼠中, 雷帕霉素能降低脂肪组织量.因此,靶向抑制mTOR/S6K1/4E-BP可预防肥胖的发生.激活AMPK能抑制mTOR,促进骨骼肌中的葡萄糖摄取和脂肪酸氧化,抑制肝脏中脂质和葡萄糖的合成,同时促进脂质氧化,降低机体葡萄糖和脂质水平,减少脂质异位沉积,增强胰岛素敏感性,抑制摄食行为的发生,为肥胖的治疗提供了新的切入点[122-124].
5 细胞代谢网络研究的瓶颈及努力方向
细胞能量代谢网络由千千万万的生化反应组成,这些反应归属于众多的细胞代谢通路.细胞能量代谢的复杂性还体现在很多代谢通路都是两用代谢通路,且不同通路间通过交叉对话彼此联系、互相制约.细胞根据能量状态及内环境的改变及时调整能量代谢网络的运转,使其能随时满足细胞对物质及能量代谢的要求;而要实现这个目标,细胞必须依赖于细胞能量代谢信号网络的正常运行.目前,人们对细胞能量代谢信号网络还缺乏规律性和系统性的认识.为了更好地认识这一信号网络的运行规律,首先要了解其部分特点.细胞能量代谢信号网络对能量代谢的调控有以下特点:1)区域化,真核细胞在进化过程中出现了不同的细胞器,将细胞分割成不同的空间,使同一代谢通路往往在特定的细胞器中进行,从而提高化学反应及调控的效率.更有甚者,一种细胞器的不同部位(亚细胞器结构)为不同的信号通路提供场所,如糖酵解在细胞质中进行,TCA循环在线粒体基质中进行,而氧化磷酸化则在线粒体膜上进行.2)远程调控,如胰岛素激活其受体发生在细胞膜上,而其下游mTORC1的激活发生在晚期溶酶体表面,mTORC1激活S6K1及eIF4E引起的核糖体及蛋白质的合成则发生在核糖体上.3)多蛋白复合体,为了能及时、准确地调控能量代谢,起“开关”作用的关键调节分子如AMPK、mTOR等往往形成巨大的多蛋白复合体,通过复合体组分的动态变化实现代谢通路的启动或关闭.4)调节分子多样化,除蛋白分子外,代谢的中间产物如AMP、F-2,6-P,甚至营养物质如葡萄糖、亮氨酸、精氨酸、脂肪酸本身,都能作为信号分子与相应的蛋白分子结合,参与能量代谢的调控.5)代谢酶的多功能性,经典的代谢酶反过来调控信号网络中调节蛋白的活性,如糖酵解中的PKM2可以转录激活c-MYC的表达,而c-MYC则可进一步激活一系列参与糖酵解及生物合成的酶的表达.IDH1的R132H和IDH2的R172K突变体能产生高浓度的2-羟基戊二酸(2-hydroxylglutarate,2-HG),进而上调HIF-1α的表达.6)动态,根据细胞内环境及能量状态的变化,细胞能量代谢信号调控网络的各成分在时空上都在发生着动态变化.
基于以上细胞能量代谢信号调控网络的特点,目前的研究瓶颈和未来可能努力的方向有:
她身材高挑,留着修剪精细的短发,穿着一件质地很好的灰色羊绒衫,脸色有些苍白。老福心想,她虽然不算是个美人,但气质独特,特别是她盯着人看的那双眼睛,成熟而内敛,让老福感到了紧张。
1)原位
目前对代谢的研究集中在体外培养细胞和离体组织.体外培养细胞的培养环境不能很好地反映机体内环境的真实情况;离体组织在处理过程中,迅速的缺血、缺氧等变化会导致细胞代谢发生剧烈变化,也很难反映动物体内活体组织细胞在原位的代谢变化,因此急需开发新的代谢物标记技术、蛋白和代谢物的原位无损检测及微量检测技术.
2)实时动态
目前的各种检查技术,包括组学和对代谢物的检测,都是确定组织或细胞在某一时间点的各种生物分子的相对数量,很难对同一组织或细胞进行动态检测,了解各种生物分子在某一生命过程中的动态变化.亟待开发相应的技术动态研究同一细胞、组织在不同生理情况下或不同发展阶段的代谢变化,如细胞在分化、衰老、恶性转化过程中的代谢变化,以及组织在不同生理、病理情况下的代谢变化.值得一提的是,最近Deplancke课题组[125]建立了一种利用射流力显微镜在RNA提取过程中保持细胞活力的单细胞转录组分析方法——Live-seq,通过在不同的时间点对同一细胞进行单细胞转录组分析和下游分析,开辟了单细胞瞬时动态分析的新领域.该技术有望拓展至质谱分析,检测蛋白和代谢产物.
3)区域化
在组织器官层面,由于组织器官内部不同区域存在结构和功能的差异,如肾脏的皮质和髓质、垂体的前叶和后叶、肿瘤的中心和四周等,目前尚缺乏对不同区域的原位代谢研究方法,开发这类研究方法也是下一步代谢研究的发展方向.
在细胞层面,不同亚细胞结构和区域的代谢差异很大,目前尽管可以借助超高分辨荧光显微技术了解细胞能量代谢信号调控网络的各组分在不同生理状态下的空间(亚细胞、亚细胞器)分布的变化规律,借助质谱技术了解细胞能量代谢信号调控网络中各组分含量在不同空间(细胞器)及多蛋白复合体中的变化规律,但是仍然缺乏检测各种细胞器中代谢调控分子和代谢中间产物的方便、快捷的技术和方法,缺乏在活的单细胞水平上动态检查代谢变化的技术和方法.
4)定量
目前对代谢调控分子和代谢中间产物的研究主要以定性描述为主,极其缺乏定量检测的方法和手段.如缺乏定量测定活体组织内部不同区域在不同生理、病理情况下代谢调控分子和代谢中间产物的技术和方法,同样尚不清楚各种细胞器内代谢调控分子和代谢中间产物的绝对定量.因此,代谢的定量研究任重道远,是今后需要努力的方向之一.
5)多参数
代谢网络极其复杂,要揭示某一特定代谢产物的变化规律,必须同时考虑时间因素、空间因素、组织器官间的交流、细胞器间的交流等问题,必须同时具备对生物大分子如核酸、蛋白及代谢小分子的检测方法.因此,测定多参数的多种方法的联合应用也是未来的一个发展方向.
总之,只有系统地揭示细胞能量代谢信号调控网络的整体运行规律,才能为癌症、肥胖、糖尿病、心血管疾病等因细胞能量代谢紊乱而引起的疾病的预防及治疗提供理论基础及药物靶点,最终为提高人类的健康水平和生活质量贡献力量.
