单磷酸腺苷激活的蛋白激酶(AMPK):能量、葡萄糖感受器和代谢性疾病治疗靶标
2022-05-20张宸崧王子涵陈燕雯万芷辰王心茗陈思薇崔立枫
张宸崧,王子涵,陈燕雯,万芷辰,王心茗,陈思薇,崔立枫
(厦门大学生命科学学院,细胞应激生物学国家重点实验室,福建 厦门 361102)
单磷酸腺苷激活的蛋白激酶(AMPK)是一种重要的调节细胞物质和能量代谢的蛋白质.早在1973年,人们便发现从大鼠肝脏中提取出来的一个具有激酶活性的组分,同时具有磷酸化并抑制乙酰辅酶A羧化酶(ACC)和3-羟基-3-甲基戊二酸单酰辅酶A还原酶(HMGCR)这两个脂质合成关键蛋白的活性[1-2],后续研究表明该组分能够被单磷酸腺苷(AMP)所活化[3-4].鉴于此,在1988年它被重新命名为AMPK[5].AMPK能通过磷酸化其下游的多种蛋白达到促进分解代谢、抑制合成代谢的效果,从而在生物体内发挥在能量水平上“开源节流”的作用.本文从AMPK的两类调控方式入手,详述其在代谢性疾病上“纠偏”的作用,并解释其生理功能和作为药物靶标的潜力.
1 AMPK的结构
如图1所示,AMPK是一个异源三聚体,由α、β和γ 3种亚基组成.在哺乳动物中,α和β亚基分别有2种亚型[6-7],γ亚基有3种亚型[8],这就意味着理论上有至少12种AMPK蛋白结构.最新的研究表明,AMPK的3种亚基的不同亚型在表达和调控方式上具有时空特异性,这提示在不同的组织器官中,AMPK在调节方式和下游效应上可能存在着差别[9-10],也提示以AMPK为靶点的药物开发可能需要注意这种特异性[11].
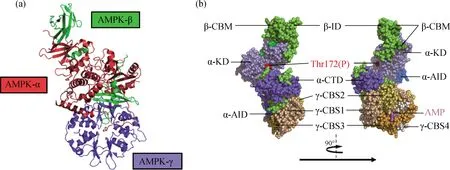
图(a)数据来自PDB 4cff;图(b)数据来自PDB 6c9j;KD.激酶结构域;AID.自抑制结构域;CTD.羧基端(C端)结构域;CBM.碳水化合物结合模块;ID.交互结构域;CBS.胱硫醚β合成酶序列;Thr172(P).苏氨酸172(磷酸化).
AMPK的α亚基是重要的激酶活性亚基,主要由α-KD、α-AID和α-CTD组成.α-AID的存在会抑制α-KD 的激酶活性[12];在α-KD的C端小叶上有一个重要的位点——Thr172,AMPK的上游激酶对该位点的磷酸化能够解除α-AID的自抑制作用,是最大程度地激发AMPK活性所必需的[12-13].
AMPK的β亚基结构尚未完全解析,主要包括氨基端(N端)的豆蔻酰化区域、β-CBM和β-ID.β亚基N端的豆蔻酰化修饰对AMP和二磷酸腺苷(ADP)促进AMPK的Thr172位点磷酸化有关键作用[14-15],而β-CBM本身则能和糖原等营养物质结合并调节AMPK[16].CBM还能和α亚基共同作用,在交界处形成变构药物和代谢物(ADaM)位点,是重要的药物靶向变构激活AMPK的位点[17].
AMPK的γ亚基含有4个CBS串联重复序列,可以与AMP、ADP或三磷酸腺苷(ATP)结合,影响AMPK的构象.关于AMPK结构的更多细节和最新进展可参阅相关综述文献[18].
2 AMPK的调控机制
2.1 经典机制:通过AMP/ATP比值调控的变构激活
如上所述,AMPK发现的过程是和细胞内低能量水平的表征分子AMP紧密联系在一起的,因此和AMP等腺苷类物质相关的激活被称为经典机制.在细胞中,ATP是生物体内能量的“通货”,ADP是ATP供能后失去一个磷酸基团的产物,AMP则是ATP失去一个焦磷酸基团或ADP失去一个磷酸基团的产物.在一般生理条件下,这3种物质的比例处于一种比较稳定的状态;而当机体处于过度饥饿、缺血或者其他极端条件下时,ATP产生不足而ADP累积,此时机体通过2ADP → ATP + AMP反应在一定程度上弥补ATP的不足,因而导致AMP的水平升高[19].AMPK正是通过其γ亚基上的腺苷酸结合位点来感应这3种腺苷酸的水平:该位点由4个高度相似的CBS区段——CBS1、CBS2、CBS3和CBS4组成,除CBS2外,另3个区段都可以结合腺苷酸,且CBS1和CBS4对腺苷酸的结合还能加强CBS3结合AMP的能力[20],使AMPK感应AMP的能力较ADP和ATP灵敏得多[21].当机体AMP含量较高,即能量水平较低时,CBS3与AMP结合,使γ亚基发生变构,这种变构调节导致α亚基的Thr172位点暴露,进而促进AMPK的上游激酶对其的磷酸化,解除AID对KD的抑制作用,从而提高AMPK的活性[22-23].除AMP外,ADP也能结合CBS3,但其作用尚存在争议[15,21,24];而当机体内ATP含量较高即能量水平较高时,CBS3则被ATP占据,使AMPK保持在利于Thr172位点被去磷酸化的构象,引起其活性的抑制[20,25].AMPK正是借此感应细胞内的能量水平,并在低能量水平下被激活,进而调节一系列下游信号通路,抑制合成代谢、促进分解代谢,以维持正常的机体能量水平.生化实验证据显示AMP/ATP比值随ADP/ATP比值的平方而变化[19],故而感知AMP相较于ADP更灵敏.加之不同γ亚基的亚型感应AMP的能力不同,如在体外γ1和γ2感应AMP的能力显著强于γ3[26],且如前所述不同亚型的组织和细胞的时空特异性分布不同,这些机制很可能保证了细胞内的能量变化被灵敏地感知并被严谨地调控.
体外实验结果表明,Thr172位点的磷酸化可使AMPK活性提升100倍以上[27],因此被认为是AMPK最重要的活性调节方式.目前已发现的可以磷酸化Thr172位点的上游激酶主要有3种,其中和感应能量水平即AMP、ADP调控相关的是肝脏激酶B1(LKB1).它于1998年在波伊茨-耶格综合征中被发现[28],并随后被证明对Thr172位点的磷酸化起主要作用[29-31].尽管LKB1和AMPK是激酶与底物的关系,但AMP或ADP促进的LKB1对AMPK的磷酸化却不仅需要这两个分子本身,还需要更多胞内结构的参与:原核表达的AMPK在体外(游离状态)无法重现AMP、ADP的这一作用[32].而在1997年,Mitchelhill等[33]发现AMPK的β亚基N端能被豆蔻酰化修饰,提示AMPK在胞内是能够结合到细胞膜系统上的;2010年,Oakhill等[14]进一步发现,β亚基N端第2位甘氨酸位点的豆蔻酰化修饰能够促进AMPK结合到膜上,且这是AMP、ADP激活AMPK所必需的:通过对原核表达的AMPK进行豆蔻酰化修饰,就能使其响应AMP、ADP,促进LKB1对Thr172位点的磷酸化;此外,使用依赖于糖酵解产能(葡萄糖饥饿后能引起AMP升高)的COS7细胞系,在葡萄糖饥饿的条件下,在细胞水平验证了AMPK膜定位的重要性.
AMP对AMPK的变构激活还体现在其能使被磷酸化后的AMPK活性进一步提升2~3倍(随ATP水平而变[21]),达到最大程度的激活[34].AMP和ADP的结合还有抑制AMPK被磷酸酶去磷酸化的作用[24,35],最新研究表明这其中还需要多个分子调节AMPK出入细胞核[36].
AMPK的变构调节机制是设计靶向AMPK的小分子药物的重要依据.以MK-8722(靶向ADaM位点)[37]等药物为代表的AMPK变构调节激活剂[38],被证明可以有效避免基于抑制线粒体ATP产生以提高AMP水平的机制所设计的药物导致的细胞毒性[39-40],因而具有很好的应用前景;但这类化合物目前需要解决的关键问题是如何避免无差别、强烈地全局性激活体内AMPK所引起的副作用.
2.2 非经典机制:通过钙离子或对葡萄糖水平感应的激活
除依赖AMP、ADP之外的AMPK激活方式被统称为非经典机制.其中,钙调素依赖蛋白激酶的激酶β(CaMKKβ)能被胞内大量钙离子水平升高所激活(通过结合钙调蛋白,后者在钙离子结合的情况下结合CaMKKβ,使其自抑制作用被解除),再引起AMPK的Thr172位点磷酸化,因而在神经等系统中发挥重要作用[41].DNA损伤所引起的AMPK激活也被认为由CaMKKβ介导[42].而转化生长因子β活化激酶1(TAK1)则是一种有丝分裂原活化蛋白激酶的激酶(MAPKK),在内吞体/溶酶体损伤、有丝分裂原刺激等条件下能够引起AMPK的激活[43-44].现有证据表明,至少在体外的单一体系下,CaMKKβ和TAK1对AMPK的激活独立于AMP依赖的AMPK激活途径[44-45],尽管在特定的生理条件下两者之间可以交互影响[43].除CaMKKβ和TAK1介导的非经典激活外,靶向ADaM位点的药物如MK-8722、水杨酸和A769662,则能够通过抑制Thr172位点的去磷酸化以及变构调节的方式激活AMPK[46-48].
AMPK的另一类非经典激活方式则与机体感应葡萄糖水平的降低有关,它是AMPK激活最普遍的情况,不仅常见于如餐间饥饿、运动等多种生理条件下,而且从酵母到哺乳动物高度保守[49].尽管葡萄糖本身作为机体主要的能量来源和ATP的产生有密切联系,但与COS7细胞系以及许多肿瘤细胞系不同,本课题组及相关研究都发现,在肌肉、肝脏等不依赖于糖酵解为主要ATP生产方式的正常组织中,葡萄糖水平的下降并不能引起AMP水平的上升[50-51],这很可能是由于机体和组织内丰富的氨基酸(如谷氨酰胺)、脂肪酸等替代碳源能够在葡萄糖水平下降时及时补充并生产ATP[52].本课题组发现:此时AMPK的激活需要在溶酶体膜表面进行,依赖于体轴发育抑制因子(AXIN)、定位在溶酶体膜上的Ragulator复合体和v型ATP酶(v-ATPase)的参与.当葡萄糖水平下降时,v-ATPase被抑制,它和Ragulator复合体发生构象变化,促进AXIN与之结合并被招募到溶酶体附近.AXIN则可以结合LKB1,作为“桥梁”连接定位在溶酶体附近的AMPK和LKB1,从而促进LKB1对Thr172位点的磷酸化,最终在溶酶体附近激活AMPK[53-54].早在这一机制发现之前,已有研究通过蛋白质组学手段鉴定AMPK能定位在溶酶体上[55],且本课题组发现的AMPK激活机制也依赖于β亚基的豆蔻酰化,因此将上述机制称为“溶酶体途径”[54](图2).需要注意的是,相较于豆蔻酰化,尽管β亚基的CBM与糖原对AMPK的调节有关,但却并不参与葡萄糖对AMPK的调节[56],ADaM位点也不参与该过程[51].有意思的是,Ragulator早先被认为是细胞内另一个重要代谢调节者——雷帕霉素靶蛋白复合体1(TORC1,在哺乳动物中称为mTORC1)维持活力所必需的[57-58];与AMPK相反,mTORC1在细胞内能量和物质充足时迁移到溶酶体表面被激活并启动合成代谢,促进细胞增长和增殖[59].可见AMPK和mTORC1的激活对应于不同的营养、能量条件,却“借道”同一类分子行使调控作用,这可能是一种简约而又精确的调节方式.
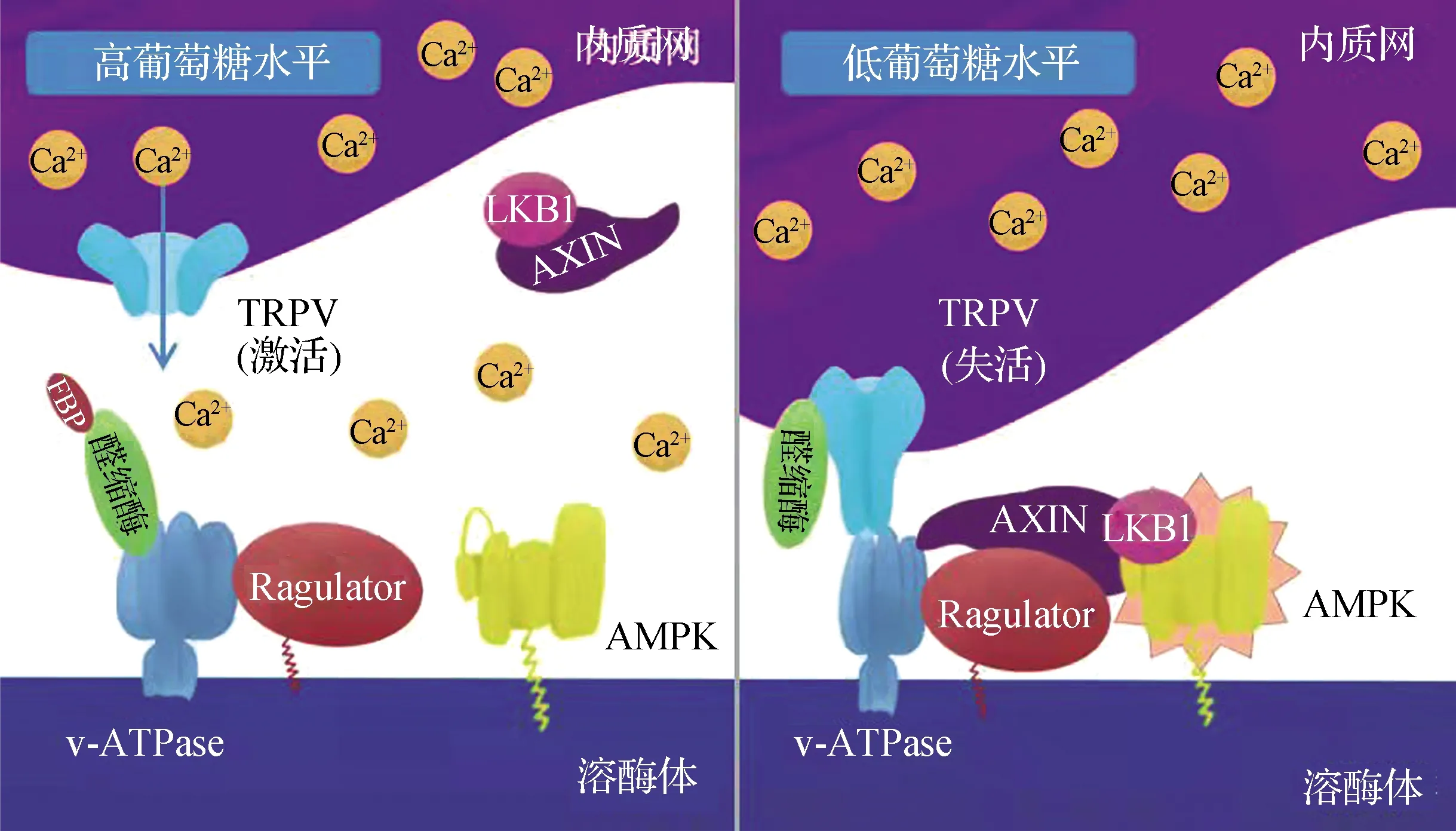
FBP.果糖-1,6-二磷酸.
葡萄糖水平下降能够在不依赖AMP的基础上激活AMPK,这说明它有另一套信号被感知以调控AMPK.本课题组发现糖酵解途径的中间产物FBP是介导这一过程的关键信号分子[51].FBP在葡萄糖水平下降时下降,不再结合催化它形成三碳糖的醛缩酶,进一步导致v-ATPase-Ragulator复合体的变构[60](图2),可见醛缩酶是感受FBP乃至葡萄糖水平的感受器.作为糖酵解途径最直接(没有旁路)的产物之一,FBP的产率常被认为是糖酵解速率的表征[61],因此它比AMP更灵敏,在细胞AMP水平升高前AMPK即可通过感知机体葡萄糖水平的降低而被激活,“前瞻性”地调节细胞代谢稳态.有意思的是,本课题组在体外单纯含有v-ATPase和醛缩酶的体系中,无法观察到FBP对v-ATPase的调节,这强烈暗示还有其他因子参与该过程;进一步鉴定发现阳离子通道瞬时受体电位香草酸亚型(TRPV)是介导这一过程的关键.TRPV主要定位于细胞膜和内质网上,其中有一部分内质网定位的TRPV能够在内质网-溶酶体接触的位置和溶酶体途径产生联系:低葡萄糖水平下,未结合FBP的醛缩酶结合并抑制TRPV的活性,抑制其从内质网释放钙离子的作用,从而降低内质网-溶酶体接触位置的钙离子浓度,使得TRPV结合v-ATPase从而抑制其活性[60](图2).需要注意的是,这里所提到的钙离子,并非是前述能激活CaMKKβ所释放的大量钙,而是原本很低浓度的钙离子浓度进一步降低所引起的情况,对AMPK的调节作用也并不同于前者.
在分子机制上,溶酶体途径和经典机制是相互独立的,即AMP可以绕过溶酶体途径对AMPK行使激活的功能,而溶酶体途径可以不需要AMPK结合AMP,两者并不互相影响;在层级关系上,经典机制又是溶酶体途径的上级,能造成比溶酶体途径更强、更广泛的AMPK激活.这进一步体现在不同激活方式下AMPK所具有的时空特异性调控特点上:当葡萄糖浓度下降(AMP水平还未上升)时,溶酶体上的AMPK先经溶酶体途径被激活;当AMP水平上升时,如前述更加严重的营养缺失发生时,胞质中的AMPK也开始逐渐被激活,此时AXIN通过直接加强LKB1和AMPK的相互作用介导该过程[53];而当细胞中能量严重匮乏,如在缺氧、缺血等情况发生时,线粒体上的AMPK最终被激活,这只需要AMP的参与.鉴于能量严重匮乏的情况并非生理常态,该机制可能部分解释了为何直接靶向AMPK,以及以引起全局性AMPK激活为策略所设计的药物往往难以避免一些副作用,如MK-8722所引起的心肌肥大[37],后者最近被证实确实是由于心脏中AMPK过度激活所导致的[62].因此,基于葡萄糖感应的溶酶体途径也许可为针对不同代谢疾病设计相应的引起AMPK激活的药物提供新思路.
3 AMPK的底物及其对代谢的调控
如上所述,AMPK在功能上可以概括为“促进分解代谢,抑制合成代谢”,从而在营养和能量匮乏的情况下维持机体的物质与能量稳态.AMPK通过磷酸化一系列底物直接或间接行使这些功能[63](图3).
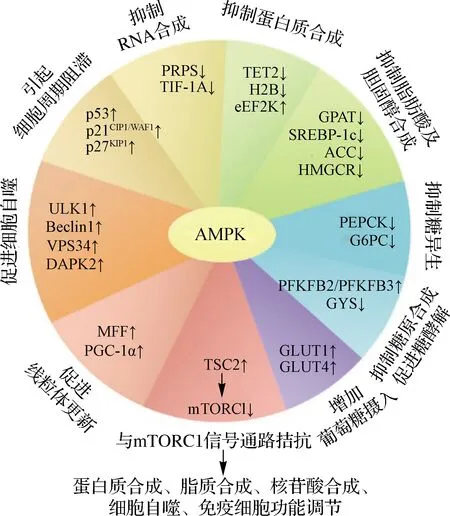
TET2.Tet甲基胞嘧啶双加氧酶2;H2B.组蛋白2B;eEF2K.真核延伸因子2激酶;GPAT.甘油-3-磷酸酰基转移酶;SREBP-1c.固醇调节元件结合蛋白-1c;PEPCK.磷酸烯醇式丙酮酸羧激酶;G6PC.葡萄糖-6-磷酸酶催化亚基;PFKFB.6-磷酸果糖-2-激酶/果糖-2,6-二磷酸酶;GYS.糖原合酶;GLUT.葡萄糖转运蛋白;TSC2.结节性硬化症复合体亚基2;MFF.线粒体裂殖因子;PGC-1α.过氧化物酶体增殖物激活受体-γ共激活因子-1α;ULK1.Unc-51样自噬启动激酶1;Beclin1.自噬效应蛋白1;VPS34.分拣蛋白34;DAPK2.死亡相关蛋白激酶2;PRPS.磷酸核糖焦磷酸激酶;TIF-1A.转录中介因子-1α;p53、p21CIP1/WAF1、p27KIP1均为细胞命运决定相关蛋白.
3.1 AMPK调节营养物质代谢
AMPK对糖、脂质、核苷酸和蛋白质等已知的所有种类的营养物质都具有调节作用.在糖代谢方面,AMPK促进葡萄糖的分解,抑制葡萄糖的生成和储存.AMPK可以磷酸化并抑制GYS,抑制糖原合成[64-65];也可磷酸化磷酸果糖激酶PFKFB2和PFKFB3,促进糖酵解[66-67];还可以促进葡萄糖转运至细胞内以维持其分解,如分别通过磷酸化Tre-2/USP6-BUB2-cdc16结构域家族成员1(TBC1D1)和硫氧还蛋白互作蛋白(TXNIP),促进GLUT4和GLUT1转移到细胞膜上[68-69],或通过转录调节因子肌细胞增强因子2(MEF2)和组蛋白脱乙酰基酶5(HDAC5)提升GLUT4的表达水平[70-71].在促进糖酵解的同时,AMPK也抑制糖异生作用,如通过磷酸化初乳碱性蛋白(CBP)、环磷腺苷效应元件结合蛋白调控的转录共激活因子2(CRTC2)、HDACs(及其下游的叉头框蛋白O(FOXO))、碳水化合物反应元件结合蛋白(ChREBP)等一系列转录因子和转录调节因子调节其转录活性,或直接调节干细胞核因子4α(HNF4α)等转录因子的表达水平,抑制PEPCK、G6PC等糖异生相关限速酶的表达[72-76].需要注意的是,AMPK相关激酶(ARK)家族的其他蛋白也能行使AMPK抑制糖异生的功能(如对CRTC2和HDACs的磷酸化)[77],从而保证糖异生这一高度耗能的过程在营养和能量水平较低时被准确地调控.
脂质合成是高度耗能的代谢过程,而它本身作为机体重要的储能物质,则能够被分解而产生大量ATP.作为其经典功能之一,AMPK通过多个底物促进脂质的摄取和分解,抑制脂质合成.AMPK可以磷酸化并抑制ACC(包括定位于细胞质的ACC1[1]和线粒体的ACC2[78])的活性,减少丙二酸单酰辅酶A的产生,而后者的产生既是脂质从头合成(DNL)途径的限速步骤,本身又能够抑制线粒体进行脂肪酸的摄取和β-氧化[1,79].同样地,AMPK对HMGCR的磷酸化和抑制也是抑制胆固醇合成的关键[2].与糖代谢类似,AMPK还能促进脂肪酸转运体CD36向细胞质膜的转移,促进细胞摄入脂肪酸[80].AMPK还可以磷酸化转录因子SREBP-1c并抑制其被蛋白酶降解,后者是调控包括ACC、脂肪酸合成酶(FAS)等DNL关键基因转录的因子[81].类似地,AMPK也能通过上述磷酸化ChREBP的机制,从转录水平抑制DNL[82].除脂肪酸合成和β-氧化外,AMPK还能通过磷酸化GPAT,抑制甘油三酯(TAG)和磷脂合成的共同底物——甘油二酯(DAG)的生成,从而抑制TAG和磷脂的合成[83].
尽管被认识得较晚,但已有多项研究表明,与糖类、脂质类似,核酸也是重要的储能物质,且其合成也是高度耗能的.如在胸腺细胞中,RNA的合成至少消耗了15%的有氧呼吸产能[84],而作为细胞内核酸的储存库之一,核糖体(储存体内80%以上的核糖体RNA)能够在营养物质缺乏时被降解以产能[85].现有研究表明AMPK能够抑制核酸的合成,如:AMPK能磷酸化并抑制嘌呤合成途径的限速酶PRPS,从而抑制嘌呤核苷酸的从头合成[86];AMPK也能磷酸化并抑制调控RNA聚合酶Ⅰ转录的关键转录因子TIF-1A,从而抑制RNA的合成[87].
蛋白质的合成也是机体内主要的耗能过程,类似于核酸,胸腺细胞中的蛋白合成消耗了约20%的有氧呼吸产能[84],AMPK则能通过多种方式抑制蛋白质的合成,促进蛋白质的分解.AMPK最主要的作用是通过抑制mTORC1阻止蛋白质的翻译起始:AMPK能够磷酸化TSC2并激活TSC1/TSC2/TBC1D7复合体,后者作为mTORC1的激活因子——小G蛋白Rheb的三磷酸鸟苷酶活化蛋白,能够直接抑制mTORC1活性[88-89];AMPK也可以磷酸化mTORC1复合体上的Raptor亚基从而直接抑制mTORC1活性[90].需要注意的是,除促进蛋白质的翻译外,mTORC1也能促进脂质(如SREBP1[91]和HMGCR[92])和核苷酸(包括嘌呤[93]和嘧啶[94-95])的合成,因此AMPK通过mTORC1调节机体代谢的方式可谓“牵一发而动全身”.另外还需要说明的是,和前文溶酶体途径中提到的AMPK与mTORC1响应葡萄糖水平并经共同因子在溶酶体上被调控的作用不同,AMPK对于mTORC1的抑制仅限于其活性而不涉及mTORC1的溶酶体定位,它起到的是快速而灵敏地抑制mTORC1的作用:当AMPK缺失时,mTORC1仍能响应葡萄糖水平下降,离开溶酶体表面从而被抑制,但速度明显变慢[96].除mTORC1外,AMPK还可以通过磷酸化并激活eEF2K从而抑制多肽链的延伸[97],或通过磷酸化并抑制组蛋白H2B和双加氧酶TET2在转录水平上抑制蛋白的表达[98-99].
3.2 AMPK调节细胞代谢
除调节糖、脂质等具体代谢途径外,AMPK还能在细胞水平上整体性地调节代谢过程,包括促进自噬作用、维持线粒体的质量和数量以及调节细胞周期等过程.
自噬作用,特别是巨自噬作用,是细胞无差别降解生物大分子乃至包括线粒体、脂滴、核糖体、内质网等细胞结构,维持能量产生和营养物质释放与再利用的关键过程.AMPK的激活可通过以下至少4个方面的机制显著促进细胞的自噬作用:1)AMPK可以磷酸化启动自噬的关键蛋白ULK1的多个位点,激活ULK1,从而启动自噬[100-101];2)ULK1也受到mTORC1的结合与抑制,而AMPK对mTORC1的抑制能够解除mTORC1对ULK1的抑制作用,从而激活ULK1[101];3)AMPK可以磷酸化自噬调节相关蛋白Beclin1和VPS34,也可与ULK1协同调节VPS34,从而促进VPS34和Beclin1共同组成自噬启动复合体,加快自噬[101-102];4)AMPK还能够磷酸化并激活DAPK2,促进DAPK2对Beclin1的磷酸化从而促进自噬作用[103].
AMPK对线粒体的作用可归纳为“除旧迎新”,即通过清除细胞内已损坏、呼吸效率低下(如由呼吸作用所产生的活性氧(ROS)而造成膜损坏,引起质子泄漏和质子梯度部分丧失等)的线粒体,促进新的线粒体生成.在清除受损线粒体方面,AMPK可以通过加强自噬作用促进受损线粒体的降解[100];同时,AMPK能够通过磷酸化MFF促进线粒体的分裂,使得受损的线粒体更易分裂从而经自噬作用被降解[104].而在促进线粒体生成方面,AMPK能够磷酸化并激活PGC-1α的转录活性,后者是介导线粒体生成所需核编码基因的转录所必需的[105].AMPK也能够通过上调辅酶Ⅰ(NAD+)的水平,促进NAD+调节的去乙酰化酶Sirtuins对PGC-1α的去乙酰化,进一步上调PGC-1α的转录活性[106].此外,上述AMPK对合成代谢的抑制作用(如ACC调节的脂肪酸合成)在维持能量平衡的同时减少细胞内的还原力消耗,从而维持胞内ROS水平即氧化还原稳态,以保护线粒体结构和功能的完整性[107];同时,AMPK也能够通过FOXO、核因子E2相关因子2(NRF2)等转录因子,在转录水平上维持细胞中抗氧化蛋白的表达水平[108-109].特别地,AMPK还能够通过上述加强糖、脂质等分解代谢途径的方式,瞬时性地提升线粒体的有氧呼吸强度(该作用被称为“mitohormesis”),也能进一步激活PGC-1α,促进线粒体生成[110].AMPK的上述作用使得胞内线粒体具有更高的呼吸效率,在产生等量ATP的同时,减少质子的泄漏和ROS的产生.总体上看,AMPK的这一作用可促进细胞代谢模式转向有氧呼吸,这也是后文所述其抑制炎症、癌症、延长寿命等功能的重要依据[111].
此外,AMPK能引起细胞周期的阻滞,暂停细胞分裂过程中众多合成代谢过程所引起的能量消耗,例如:AMPK能够磷酸化并激活p53,通过其下游周期细胞依赖性激酶(CDK)的抑制因子p21CIP1/WAF1阻滞细胞周期于G1期[112];AMPK也能够磷酸化另一CDK的抑制因子p27KIP1,介导G1期阻滞[113];除G1期阻滞外,在成熟红细胞中还发现AMPK能够阻滞细胞周期于S期,并可诱导细胞凋亡,从而可能在维持红细胞更新的过程中发挥重要作用[114].AMPK在调节细胞周期方面的作用也是后文所述的抑制癌症相关功能的依据.
3.3 AMPK与免疫反应
近年来随着代谢与免疫的联系被不断揭示,传统上的免疫反应被更广泛地认为是一种代谢所介导的过程[115];而AMPK在这一过程中,特别是在调节炎症相关免疫反应中起到了关键作用,该作用可以看作AMPK调节一类特化细胞的代谢所介导的功能[116].目前,已发现AMPK能通过调节巨噬细胞、胸腺依赖性淋巴细胞(T细胞)、中性粒细胞和树突状细胞等免疫细胞类群,抑制炎症反应的发生和发展.
AMPK能够引起巨噬细胞从M1促炎类群转变为M2抗炎类群[117].虽然具体分子机制尚不明确,但是已有研究发现,AMPK的这一功能可能和其改变了M1巨噬细胞的代谢模式而使之接近于M2巨噬细胞有关[116].在M1巨噬细胞被活化(即“经典激活”,如经历脂多糖(LPS)或干扰素γ刺激)时,细胞的代谢模式转向有氧糖酵解并加强戊糖磷酸途径,而氧化磷酸化则被抑制且变得不完整[118-119],这一作用可能是M1巨噬细胞中促炎因子的生物合成所必需的[115](类似于后文所述的肿瘤细胞),且其产生的高浓度ROS和NO(由不完整的电子传递链介导[120-121])则进一步辅助其杀灭病原体[122-123].其中,M1巨噬细胞中PFKFB3的表达水平升高和Toll样受体4(TLR4)所激活的mTORC1有关,后者进一步引发缺氧诱导因子1α(HIF1α)的表达水平上升.HIF1α原本被发现是在缺氧的条件下稳定表达,使乳酸脱氢酶 A和磷酸肌醇依赖性激酶1(PDK1)等基因表达上调,提升糖酵解产能以缓解缺氧所引起的有氧呼吸产能不足;而在这里它则通过同样的方式促进有氧糖酵解[118,124].同时,NO的产生则破坏了电子传递链[119].如3.1小节所述,尽管AMPK能激活PFKFB3,但其对mTORC1的抑制起主导作用,因此整体来说AMPK抑制有氧糖酵解[125],而上述AMPK通过NAD+、Sirtuins和PGC-1α促进线粒体生成的作用也能够维持氧化磷酸化.此外,Sirtuins还能够去乙酰化并抑制核因子κB(NFκB)途径中的p65,阻止促炎细胞因子的转录[126].而在M2巨噬细胞被活化(即“非经典激活”,如受到白介素-4(IL-4)刺激)的过程中,氧化磷酸化水平则急剧升高[127].AMPK不仅可以促进IL-4通过信号传导及转录激活蛋白6(STAT6)介导的、由PGC-1α行使的线粒体生成,还能够通过促进脂肪酸β-氧化给M2巨噬细胞提供能量,从而促进该细胞的活化[128-129].
AMPK也能调控T细胞的命运决定.类似于巨噬细胞,T细胞的分化也伴随着代谢模式的改变.如CD8+T细胞被抗原呈递细胞(APC)结合后开始分裂,APC附近的CD8+T细胞进行有氧糖酵解,分化成效应T细胞;远端的则倾向于氧化磷酸化,分化为记忆T细胞[130-131].AMPK介导的mTORC1-HIF1α的抑制作用可抑制效应T细胞的分化而促进记忆T细胞的分化[132-133].类似地,CD4+T细胞在分化成辅助性T细胞的过程中,也需要mTORC1-HIF1α所介导的有氧糖酵解加强,从而行使类似于M1巨噬细胞的促炎作用;相反地CD4+T细胞分化成调节性T细胞则需要提升氧化磷酸化的水平[134-136],而AMPK的存在可促进调节性T细胞的形成[137].类似于后文所述的长寿相关代谢表型,T细胞分化中的这种代谢模式改变,可能与其在体内存活时间的长短以及对合成代谢的不同需求有重要联系[138].
与M1巨噬细胞类似,中性粒细胞也能在LPS等刺激下由TLR4介导促炎症过程,而AMPK的激活则可以逆转该过程[139];同样地,树突状细胞的活化和成熟也需要有氧糖酵解的加强,而AMPK则可以抑制这一过程[140].
综上,AMPK通过对免疫细胞代谢模式的调节,影响了这些细胞的分化、成熟与功能.在大多数促炎症反触发的过程中,AMPK被显著抑制(如LPS[141]),因此靶向AMPK的激活以抑制炎症反应的策略,将有助于机体抵抗和有序地清除病原体的侵染(最新进展请参阅综述文献[142]),以及以慢性炎症反应为特征之一的糖尿病、肥胖和癌症的治疗(见后文详述).
需要特别说明的是,上述3个方面的AMPK对细胞的代谢调节作用是简单基于AMPK在生理上的激活后进行的相关观察,然而正如在2.2小节中所述,AMPK的激活在生理上随着能量应激强度的升高,从葡萄糖缺乏到AMP上升,呈现出累进式、区域化的激活[143].且不同区域的AMPK激活能够引起不同底物的磷酸化,并引起不同的代谢调节过程.如ACC2和MFF在葡萄糖缺乏的情况下不被磷酸化,而只能在AMP水平升高的情况下被线粒体上的AMPK磷酸化,这暗示着线粒体上AMPK底物所介导的代谢调节过程只能发生在严重的能量匮乏情况下[143].最近,该过程也被证明存在于动物体内[144].而前述DNA损伤所引起的CaMKKβ介导的AMPK激活则仅局限于细胞核内,AMPK也不能够在此时磷酸化ACC等胞质中的蛋白,可见上述AMPK-p53所介导的细胞周期阻滞和AMPK对代谢途径的调节也是相互平行的过程[42].因此,未来对AMPK在不同时空条件下的功能进行更深入的研究,将有助于针对不同的情况设计相应的药物,以更好地干预和治疗各种代谢性疾病.
4 AMPK和代谢性疾病——短期效应
上述AMPK在代谢调控中行使维持代谢稳态的功能,对由代谢紊乱特别是合成代谢过度活化的疾病(如肥胖所引起的Ⅱ型糖尿病、脂肪肝)乃至癌症的症状缓解与预防有重要意义,也因此成为代谢性疾病治疗的靶标.相较于后文所述对寿命等相对长期的作用,AMPK对代谢性疾病的作用可归纳为它的短期效应.
4.1 AMPK与肥胖相关代谢性疾病
由营养过剩引起的肥胖及其引发的Ⅱ型糖尿病、脂肪肝等并发症,是当今最具代表性且对人类健康造成日趋严重威胁的代谢性疾病[145],而AMPK对代谢调节的功能在肥胖(包括内脏脂肪积累)到糖尿病(胰岛素抵抗)发展过程中的多个紊乱代谢活动起到“纠偏”作用.目前的观点认为,营养过剩引起的脂肪累积首先导致脂肪细胞TAG增多和肥大,引起细胞氧气通透变差且消耗增加,造成相对的缺氧状态,进而触发HIF1α的上调,导致趋化因子分泌并吸引巨噬细胞、肥大细胞的聚集与活化,增加多种促炎细胞因子的分泌[146-147],后者又激活脂肪细胞中的NFκB和c-Jun N端激酶(JNK)信号通路,导致胰岛素受体(IR)及其底物(IRS)的丝氨酸磷酸化,引发其失活和胰岛素通路的抑制[148-149],该作用进一步通过血液扩展到肝脏、肌肉等外周组织,从而引起外周胰岛素抵抗[150-152].除脂肪细胞肥大外,脂肪合成的增加也改变了内质网的脂成分,引起内质网应激,后者进一步提升局部炎症反应[153-155].同时,脂肪细胞中TAG合成的增强导致中间产物DAG水平的升高,后者引起细胞内磷酸激酶Cε(PKCε)的激活,加强IR的苏氨酸磷酸化而导致胰岛素通路的抑制(实际上该作用也发生于肝脏(即脂肪肝)和肌肉中,且在肌肉中还有PKCθ参与行使类似的作用)[156-157];TAG合成的增强也导致血液中游离脂肪酸、甘油等中间产物的升高(即高血脂症),后者作为底物,被运进外周组织,进一步加剧外周组织的TAG合成及高血脂症.此外,肝脏中的TAG合成增强还能够通过高水平的乙酰辅酶A等中间产物,变构激活多个糖异生途径的限速酶[158-159],后者与胰岛素抵抗(及其导致的肝糖原分解加强)一起引起高血糖[160].血糖水平的升高又进一步激活SREBP、ChREBP等转录因子,加强DNL和TAG堆积[161-162],也可以导致细胞内ROS的增多,进一步导致内质网应激和炎症反应[163-164].由此可见,上述AMPK对DNL、DAG等脂质合成的抑制以及对β-氧化的加强作用,能够从根本上缓解包括脂肪、肝脏和肌肉在内的外周组织器官的脂质积累[165-167];其对糖异生的抑制作用、对血液葡萄糖的摄取以及对糖酵解的促进作用,则可以有效降低血糖水平[73-74];而其对炎症的抑制作用和对细胞内氧化还原稳态的维持,则能够缓解由此产生的胰岛素抵抗[128].上述关于AMPK在肥胖相关代谢性疾病中的作用总结于图4.
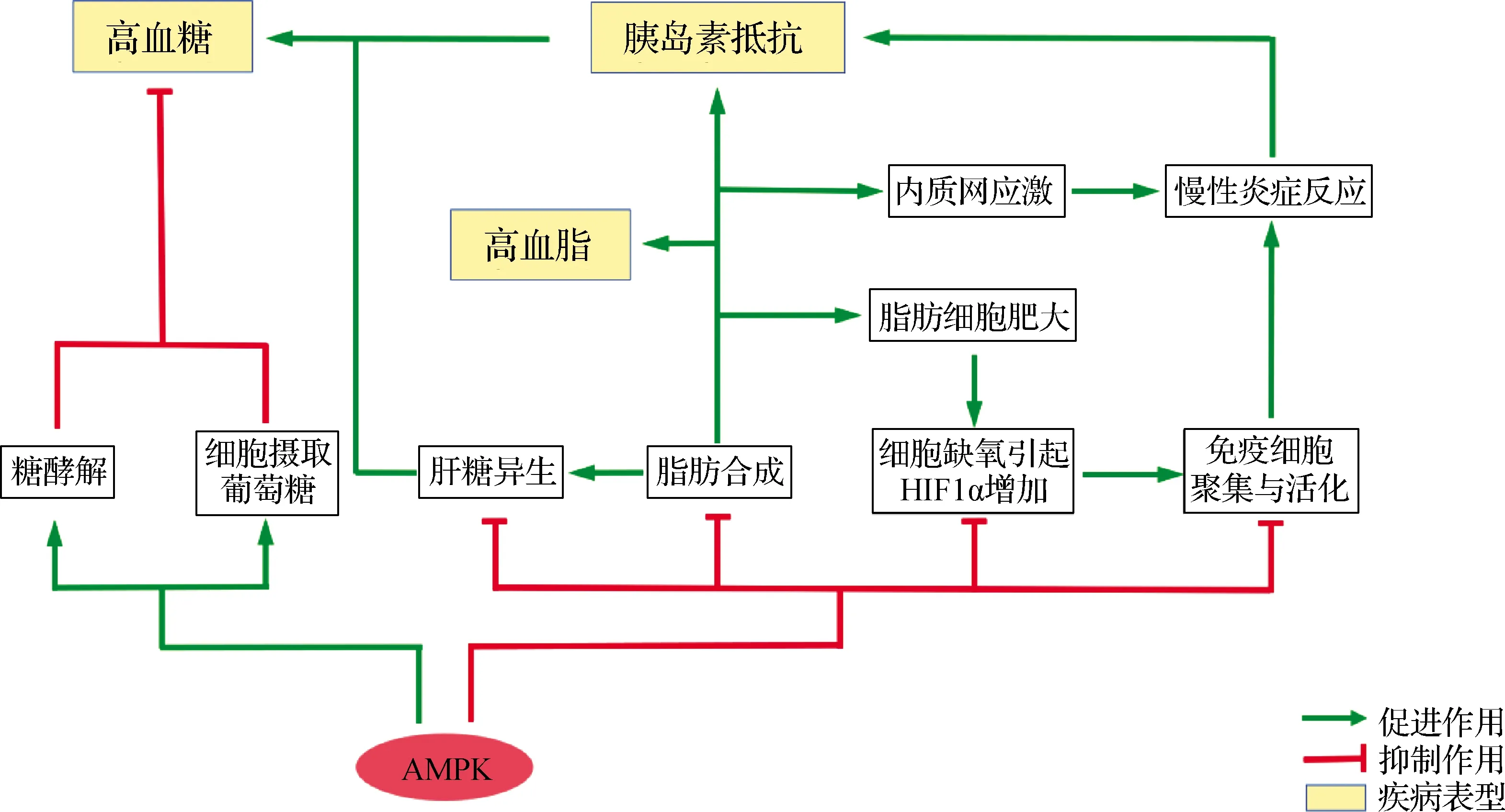
图4 AMPK在肥胖相关代谢性疾病中的作用
在动物模型和病人中血糖和游离脂肪酸水平的上升以及炎症反应的加强,能够普遍抑制AMPK活性[168-170],因此通过药物重新激活AMPK可起到显著作用.事实上,以二甲双胍为代表的AMPK激活剂已经在降低血糖和脂肪肝方面获得了很好的临床疗效,但其难以进入肌肉、脂肪等组织中发挥激活AMPK的作用[171].因此,合成新的AMPK激活剂日益受到重视,能够结合并激活AMPK的直接激活剂的研发成为重要方向,如MK-8722[37]、PF-379[167]、A769662[172]、AICAR[173](但需较高剂量[174])、水杨酸/阿司匹林[175]、C2/C13[176]、PT-1[177]和991[178-179]都被证明在缓解肥胖、糖尿病或脂肪肝中发挥作用.此外,TZD[180]、黄连素[181]、白藜芦醇[182]、人参皂苷[183]、硫辛酸[184]等AMPK的间接激活剂也被证明有类似功效.如前所述,这两类化合物亟需解决的问题是过强、过广(无时空特异性)的AMPK激活所引起的副作用.特别地,AMPK已被证明对脂肪肝进一步演变成非酒精性脂肪性肝炎(NASH,是肝纤维化、肝硬化和肝细胞癌发生的前提[185])有明显抑制作用[186].鉴于目前尚无治疗NASH的药物上市,且前述对脂肪肝有效果的药物研究也仅局限于动物模型,对相关AMPK激活剂有待进一步探索和改良,使之发挥治疗脂肪肝的作用[187].可喜的是,最近有多个来源于天然产物的AMPK激动剂,如雷公藤甲素(triptolide)、β-广藿香烯(β-patchoulene)和甜菜碱(betaine)表现出强烈的缓解脂肪肝的功能[188-190];而γ-亚麻酸(γ-linolenic acid)能够通过AMPK调节自噬,抑制脂肪肝引起的肝细胞死亡,后者被认为是脂肪肝发展成为NASH的重要因素[191-193].这些新发现的药物为脂肪肝和NASH的治疗带来了曙光.
4.2 AMPK与癌症
近年来,随着对癌症病因研究的深入,癌症作为一种代谢紊乱相关(原因和/或结果)疾病的观点被逐渐接受[194].鉴于AMPK在抑制合成代谢、阻滞细胞周期等方面的功能,它也成为多种癌症治疗的潜在靶点.遗传学方面的证据显示,AMPK的上游激酶LKB1和CaMKKβ都被证明是重要的抑癌基因[195-197],尽管LKB1也能通过ARK抑制癌症[198].敲除AMPKα1(淋巴细胞中只表达AMPKα1)则会在Eu-myc B淋巴细胞癌模型中加速癌症的发展[199],类似的效果也在p53缺失的T淋巴细胞癌模型(敲除AMPKβ1)[200]、人第10号染色体缺失的磷酸酶及张力蛋白同源蛋白(PTEN)缺失的急性T淋巴细胞白血病(T-ALL)模型(敲除AMPKα1)和前列腺癌模型(敲除AMPKβ1)中被观察到[197,201],而MAGE-A3/A6、UBE2O等泛素连接酶介导的AMPKα降解,则在多种模型中促进肿瘤的发生[202-203].除在肿瘤模型水平发挥作用外,AMPK在缓解上述肥胖相关代谢疾病的的同时,也降低了由这些疾病引起的患癌风险[204],且上述AMPK的多种激活剂已被尝试用于癌症治疗,如:水杨酸和二甲双胍联用被证明对前列腺癌和肺癌有抑制作用[205],苯乙双胍能够抑制非小细胞肺癌和T-ALL的发展[201,206],而二甲双胍还可通过AMPK磷酸化程序性死亡配体1(PD-L1)促进其降解,从而加强对乳腺癌和结肠癌的治疗效果[207].针对脂质合成代谢对肿瘤细胞生长的关键作用[208-209],最近有研究显示以ACC为靶点设计的抑制剂ND-654对于肝细胞癌的发展有很好的抑制作用[210].此外,AMPK还被报道能够磷酸化并抑制诸多原癌基因以抑制肿瘤,如:BRAF,经有丝分裂原活化的细胞外信号调节激酶-细胞外调节蛋白激酶(MEK-ERK)途径[211];YAP,经Hippo途径[212];GLI1,经Hedgehog途径[213].
然而需要注意的是,由于肿瘤微环境的多样性,在许多情况下合成代谢和细胞周期的阻滞反而利于癌细胞的存活和癌症的发展.当细胞癌变刚完成但处于营养物质匮乏的环境时,AMPK反而有利于肿瘤细胞的存活和癌症的发展,如:3.2小节所述AMPK对于细胞还原力的维持,有助于肿瘤细胞渡过葡萄糖缺乏的状态[107,214];AMPK还可通过磷酸化S期激酶相关蛋白2(Skp2)并激活蛋白激酶B(AKT),抵御凋亡并促进乳腺癌细胞抵御包括化疗药物作用在内的多种环境压力[215],而AMPK对丙酮酸脱氢酶E1亚基(PDHA)的磷酸化和激活,在促进有氧呼吸的同时提升前列腺癌细胞的ATP产率,从而有利于其在迁移过程(处于多种营养缺乏环境,详见综述文献[216])中的存活[217].在小鼠模型上,与胚系敲除AMPK不同,当T-ALL、急性髓性白血病模型中淋巴癌在骨髓处刚发生时便敲除AMPK(此时癌细胞处于葡萄糖水平远低于血液的环境中),则会促进癌症的发展[218-219].因此,未来在逐步阐释癌症发展的不同阶段所经历的代谢重编程乃至微环境的营养物水平变化基础上,辅助以时空特异性激活AMPK的用药策略,将为癌症的防治带来极大帮助.
5 AMPK和长寿——长期效应
与对脂肪肝、糖尿病和癌症等代谢性疾病的短期作用相比,AMPK的长期效应主要体现在它延缓衰老、延长寿命方面的贡献.
上述AMPK对代谢性疾病的预防作用本身就能够促进健康与长寿.例如,服用二甲双胍的糖尿病患者由糖尿病并发症(包括癌症)引起的死亡风险明显降低[220-221],二甲双胍很可能通过缓解糖尿病的病症发挥作用[222].AMPK在延长寿命方面的直接作用是通过线虫(Caenorhabditiselegans)、果蝇(Drosophilamelanogaster)等模式生物的相关研究发现的,在线虫中表达有活性的AMPK,能够显著延长其寿命[223-224];在果蝇中,AMPK和LKB1的表达都可以延长其寿命[225-226].此外,多种已知可促进健康长寿的生活方式也能够通过AMPK发挥作用.例如,目前唯一在所有已验证过的动物(包括非人灵长类和人类[227-228])中都能延长寿命、提高生活质量的生活方式——卡路里限制(CR),被发现能激活AMPK[229-230],且在线虫和果蝇中已证明缺失AMPK可影响CR介导的延长寿命的效果[224-225].上述AMPK激活剂如二甲双胍和白藜芦醇,在延长寿命、延缓衰老方面的功能也已得到广泛研究:两者都能够显著延长线虫[231-232]、果蝇[232-233]和小鼠[182,234]的寿命,且在线虫和小鼠中均呈现AMPK依赖性[231,235-237].二甲双胍和白藜芦醇的上述作用模拟了CR的效果,因而被称为CR模拟物(CRM),目前,AMPK是设计CRM的核心靶点之一[238].鉴于二甲双胍广泛的临床应用(白藜芦醇的成药性仍需进一步改善[239])和已被反复证实的安全性,加之其对非糖尿病人群的患癌率有明显抑制作用[240],它在健康人群中抗衰老作用的临床研究已经开始[241].
从机制上看,AMPK的多个下游底物都和寿命有关.目前发现和寿命相关的主要信号通路有3类:Sirtuins的活化、mTORC1的抑制以及胰岛素和胰岛素类生长因子途径的抑制[242-244].上述AMPK对Sirtuins的调节[245]、对TSC和Raptor等分子的磷酸化[246]以及对CRTCs和FOXO的抑制[224,247],则分别介导了这3个过程.在细胞层面,上述AMPK在维持线粒体的数量与质量[248-249]、mitohormesis[250]、自噬作用[251-252]、蛋白合成[253]等方面的功能也被证明与寿命密切相关.类似于肥胖和糖尿病,研究表明衰老也是一种机体的慢性炎症反应,被称为“inflammaging”[254],而AMPK对免疫细胞炎症化的抑制则能延缓衰老的过程[255].细胞的衰老,特别是免疫细胞和干细胞的衰老,被认为和寿命有极大关联[256-257].尽管有研究表明细胞衰老可能有抑制癌症发生的功效[258],但衰老细胞裂解对于延长寿命、延缓衰老有重要作用[259-260].AMPK对于细胞周期的调节作用已被证明能够抑制衰老[261-262].而目前引发衰老细胞裂解最有效的化合物Fisetin[263]也被证明能激活AMPK[264].此外,生物体的节律维持对于衰老和寿命有重要影响[265],而AMPK已被证明在该过程中起关键作用[266].最后,需要特别注意的是,尽管AMPK与健康长寿之间有天然而密切的联系,但两者之间的因果关系,特别是AMPK在引起长寿的不同策略下的具体分子机制及其与长寿的直接联系,尚未完全阐明.例如:和直接表达有活性的AMPK不同,二甲双胍对线虫寿命的延长不依赖于FOXO,而依赖于NRF所介导的类似于mitohormesis提高有氧呼吸效率的机制[224,231];同样是二甲双胍,作用于线虫的老年期,则反而会破坏线粒体功能并引起寿命的缩短[267].这暗示AMPK对长寿的调节处于复杂的时空网络下,在“正确的时间”和“正确的地点”做“正确的事”,而进一步详细阐明其中的机制将为延长寿命、维持健康提供更多思路.
6 总结与展望
AMPK作为一种调控细胞能量代谢的重要分子,通过对能量、葡萄糖等的感知维持代谢稳态,并在多种代谢综合征乃至衰老中行使重要的功能,为人类最终战胜代谢性疾病带来了信心,也因此成为重要的药物靶点.然而如本文所述,由于AMPK激活和功能的时空网络的复杂性,靶向AMPK治疗代谢性疾病的策略尚存在一定困难.今后,围绕着AMPK调节和功能上的“时空性”开展日益深入的研究,将多维度展示这一代谢性疾病治疗的靶标作用机制和功能,为最终通过操控AMPK以促进人类的健康长寿做出贡献.
致谢:感谢导师林圣彩院士和为AMPK溶酶体途径的发现做出贡献的所有前辈和后辈,还要感谢厦门大学生命科学学院的培养,并感谢厦门大学《生物学实践》课程和参与此课程的“追糖溯源”实践队全体成员为本文最终成形做出的贡献.
