5-五氟苯基-15-位阻芳基取代卟啉的水相合成研究
2022-04-01闫晓旺马晓伟顾承志
闫晓旺,马晓伟,顾承志
(石河子大学化学化工学院/新疆兵团化工绿色过程重点实验室,新疆 石河子 832003)
合成金属卟啉作为细胞色素P450的模拟酶应用于仿生催化一直是人们的关注焦点之一[1-8],不同取代卟啉模式在其催化活性方面表现各异,尤其是引入强吸电子基团的非对称卟啉备受瞩目,而这需要非对称卟啉合成化学有较大的突破[9],与经典的Alder法[10]和Lindsey[11]法在酸性体系条件下构建卟啉不同,1997年LINDSEY J S课题组研究了在碱性条件下通过四吡咯烷构建meso位四取代卟啉的合成方法,并且卟啉的分离收率可达60%[12],这有效促进了卟啉合成化学的进展,然而,LINDSEY J S课题组构建卟啉的中间体四吡咯烷(bilane)是在有机相体系中通过复杂的多步反应实现,反应条件相当苛刻,涉及到精确的无水无氧操作。EM T等[13]研究表明meso位没有任何取代基的线性四吡咯烷不稳定,对光热都很敏感,自身容易发生氧化降解或者其他耦合反应。综上所述,探究简单的可规模合成稳定的四吡咯烷用于构建卟啉仍是当前卟啉化学挑战之一。
近年来,水相合成反应也受到越来越多的关注,KOSZARNA B等[14]进行了在水-甲醇共溶剂体系中meso位取代的咔咯啉(Corrole)合成工作,从咔咯啉结构逆合成分析看,通过四吡咯烷中间体氧化关环是咔咯啉的核心反应过程,但没有给出四吡咯结构的表征。本研究课题组首次进行了水相中5,10,15-三芳基取代四吡咯烷的合成工作[15-16],较早之前ROSE S等研究发现在生物合成卟啉的过程中四吡咯烷是其合成前体[17]。本研究课题组设计在水相中通过四吡咯烷和另一分子的芳醛在酸催化下缩合生成卟啉原,再经四氯苯醌氧化生成卟啉分子。从卟啉生源合成的角度来看,作为卟啉生成过程中核心中间体四吡咯烷,必须要在水中有一定的溶解度,所以只要找到一个合适的共溶剂,就能够保证酸催化下四吡咯烷与芳醛缩合生成卟啉原,去模拟生源性卟啉合成。本文进行以上设想的初步研究,整个设计合成思路如图1所示。

图1 Trans-AB卟啉的合成
首先在水中合适浓度盐酸催化下吡咯与甲醛缩合得到二吡咯甲烷,接着在合适比例共溶剂中合适浓度盐酸催化二吡咯甲烷与五氟苯甲醛缩合生成10-五氟苯基四吡咯烷(引入强吸电子基团目的是提高四吡咯烷的化学稳定性),再在合适比例共溶剂中合适浓度盐酸催化10-五氟苯基四吡咯烷与另一分子芳醛缩合生成卟啉原,然后氯仿萃取,无水硫酸镁干燥,氯仿溶液中加入四氯苯醌(TCQ)回流氧化得到5-五氟苯基-15-芳基取代卟啉。
1 实验部分
1.1 仪器与试剂
IKA 加热磁力搅拌器,IKA 机械搅拌器,BrukerAvanceⅢ 400 HD核磁共振仪,Thermo Fisher Scientific LTQ FTICR-MS高分辨质谱仪,薄层色谱TLC板购买自青岛海洋化工厂;300~400 目柱层析硅胶购买自上海泰坦科技有限公司;其它化学试剂均为分析纯,购于上海泰坦科技有限公司和北京百灵威科技有限公司。
1.2 实验步骤及产物的结构表征
1.2.1 二吡咯甲烷的合成
在250 mL单口圆底烧瓶中,依次加入1 mL 37% HCl,100 mL蒸馏水,磁力搅拌下加入4.2 mL(56.7 mmol)新蒸吡咯,然后将0.75 mL (9.6 mmol)甲醛水溶液缓慢滴入圆底烧瓶中,室温反应30 min后,加入氨水中和至pH到9左右。用乙酸乙酯(150 mL ×3)萃取,分离有机相并用无水硫酸钠充分干燥,过滤,滤液减压浓缩,粗产品柱层析分离(石油醚∶二氯甲烷:乙酸乙酯=167∶37∶1,v/v/v作为淋洗液),Rf=0.44(石油醚:二氯甲烷:乙酸乙酯=7∶27∶1,v/v/v)。收集目标接受液,减压旋干,固体经正戊烷反复超声洗涤,过滤,减压抽干得白色固体1化合物0.584 g,产率为40%。
1.2.2 10-五氟苯基四吡咯烷的合成
在500 mL的圆底烧瓶中将二吡咯甲烷(600 mg,4 mmol)与五氟苯甲醛(244 μL,2 mmol)溶解在200 mL 乙醇溶剂中,取10 mL 37% HCl,200 mL蒸馏水加入圆底烧瓶中,室温磁力搅拌1 h,加入氨水中和,用二氯甲烷(150 mL×3)萃取,收集有机层并用无水硫酸钠干燥,得到棕色透明溶液,过滤,浓缩,粗产品柱层析分离(石油醚∶二氯甲烷∶乙酸乙酯=16∶3∶1,v/v作为淋洗液),Rf=0.36(石油醚∶二氯甲烷∶乙酸乙酯=7∶3∶2,v/v/v),真空旋干,正戊烷反复超声洗涤,过滤抽干,得到棕灰色粉末2化合物0.818 g,产率为87.0%。
1.2.3 5-五氟苯基-15-基取代卟啉合成
在500 mL 两口圆底烧瓶中,将0.4 g (0.83 mmol)10-(五氟苯基)四吡咯烷与0.83 mmol的芳醛溶于200 mL 乙醇溶剂中,在氮气保护下,搅拌约1 min 后,加入1 mL HCl+50 mL 蒸馏水,氮气吹扫置换空气3次后,常温避光搅拌16 h。反应停止后用200 mL三氯甲烷萃取,蒸馏水(100 mL ×3),饱和的碳酸氢钠100 mL 洗涤氯仿层,有机层用无水硫酸镁干燥,过滤得到棕黑色透明溶液。向溶液中加入0.6072 g (2.49 mmol)四氯苯醌(TCQ)回流1 h后,停止反应,将所得物浓缩,硅胶柱层析分离(石油醚∶二氯甲烷=9∶1,v/v作为淋洗液),Rf=0.34(石油醚∶二氯甲烷=1∶1,v/v)真空旋干,用二氯甲烷-甲醇重结晶,真空干燥箱烘干(温度60 ℃)得到紫红色固体4a至4f化合物。
1.2.4 10-五氟苯基四吡咯烷的合成
在500 mL的圆底烧瓶中将二吡咯甲烷(600 mg,4 mmol)与五氟苯甲醛(244 μL,2 mmol)溶解在200 mL 乙醇溶剂中,取10 mL 37% HCl和200 mL蒸馏水加入圆底烧瓶中,室温磁力搅拌1 h,加入氨水中和,用二氯甲烷(150 mL×3)萃取,收集有机层并用无水硫酸钠干燥,得到棕色透明溶液,过滤,浓缩,粗产品柱层析分离(石油醚∶二氯甲烷∶乙酸乙酯=16∶3∶1,v/v作为淋洗液),Rf=0.36(石油醚∶二氯甲烷∶乙酸乙酯=7∶3∶2,v/v/v),真空旋干,正戊烷反复超声洗涤,过滤抽干,得到棕灰色粉末2化合物0.818 g,产率为87.0%。
1.2.5 5-五氟苯基-15-基取代卟啉合成
在500 mL 两口圆底烧瓶中,将0.4 g (0.83 mmol)10-(五氟苯基)四吡咯烷与0.83 mmol的芳醛溶于200 mL 乙醇溶剂中,在氮气保护下,搅拌约1 min 后,加入1 mL HCl+50 mL 蒸馏水,氮气吹扫置换空气3次后,常温避光搅拌16 h。反应停止后用200 mL三氯甲烷萃取,蒸馏水(100 mL ×3),饱和的碳酸氢钠100 mL 洗涤氯仿层,有机层用无水硫酸镁干燥,过滤得到棕黑色透明溶液。向溶液中加入0.607 2 g (2.49 mmol)四氯苯醌(TCQ)回流1 h后,停止反应,将所得物浓缩,硅胶柱层析分离(石油醚∶二氯甲烷=9∶1,v/v作为淋洗液),Rf=0.34(石油醚∶二氯甲烷=1∶1,v/v)真空旋干,用二氯甲烷-甲醇重结晶,真空干燥箱烘干(温度60 ℃)得到紫红色固体4a至4f化合物。
1.2.6 中间体和目标化合物的结构表征
二吡咯甲烷(1)[8],白色固体,0.584 g产率40.0%。1H NMR (400 MHz,CDCl3) δ 7.65 (s,2H,-NH-),6.62-6.61 (m,2H,pyrrole-H1),6.20-6.18 (m,2H,pyrrole-H2),6.07 (s,2H,pyrrole-H3),3.94 (s,2H,-CH2-)。13C NMR (100 MHz,CDCl3) δ 129.06,117.33,108.21,106.44,26.26。
10-五氟苯基四吡咯烷(2),棕灰色固体,0.818 g产率87.0%。1H NMR (400 MHz,CDCl3) δ 7.92 (s,2H,-NH-),7.82 (s,2H,-NH-),6.68 (s,2H,pyrrole-H1),6.14 (d,2H,J=2.0 Hz,pyrrole-H8),5.97 (s,2H,pyrrole-H7),5.91 (s,2H,pyrrole-H3),5.861 (s,2H,pyrrole-H2),5.73 (s,1H,meso-H10),3.92 (s,2H,meso-H5)。13C NMR (100 MHz,CDCl3) δ 146.21~145.97 (m),143.77~143.79 (m),139.10~138.85 (m),136.64~136.27 (m),129.42,128.71,127.57,117.15,108.51,107.70,106.84,106.24,33.21,26.50。19F NMR (376 MHz,CDCl3) δ -141.68 (dd,2F,J=1.5,23.31 Hz),-155.87 (t,1F,J=21.81 Hz),-161.26 (td,2F,J=8.27,22.94 Hz)。HRMS (ESI) 精确质量计算值 C25H20F5N4[M+H]+:471.16081,实验值471.16136。
5-五氟苯基-15-对甲基苯基卟啉(4a),紫色固体,0.0794 g产率16.9%。1H NMR (400 MHz,CDCl3) δ 10.34 (s,2H,meso-H),9.46 (d,2H,J=4.4 Hz,pyrrole-Hβ),9.40 (d,2H,J=4.8 Hz,pyrrole-Hβ),9.13 (d,2H,J=4.4 Hz,pyrrole-Hβ),8.97 (d,2H,J=4.8 Hz,pyrrole-Hβ),8.16 (d,2H,J=7.6 Hz,Phenyl-H),7.63 (d,2H,J=7.6 Hz,Phenyl-H),2.75 (s,3H,PhCH3),-3.12 (s,2H,-NH-)。19F NMR (376 MHz,CDCl3) δ -136.68 (dd,2F,J=9.37,25.57 Hz),-152.85 (t,1F,J=21.81 Hz),-162.16 (td,2F,J=9.02,24.82 Hz)。HRMS (ESI) 精确质量计算值 C33H20F5N4[M+H]+:567.16081,实验值567.16094。
5-(2,4,6-三甲基苯基)-15-五氟苯基卟啉 (4b) 紫色固体,0.0537 g产率10.9%。1H NMR (400 MHz,CDCl3) δ 10.31 (s,2H,meso-H),9.45 (d,2H,J=4.8 Hz,pyrrole-Hβ),9.35 (d,2H,J=4.4 Hz,pyrrole-Hβ),8.92 (d,2H,J=4.4 Hz,pyrrole-Hβ),7.34 (s,2H,Phenyl-H),2.67 (s,3H,PhCH3),1.85 (s,6H,2PhCH3),-3.09 (s,2H,-NH-)。19F NMR (376 MHz,CDCl3) δ -136.73 (dd,2F,J=9.02,26.70 Hz),-152.86 (t,1F,J=22.18 Hz),-162.19 (td,2F,J=9.02,25.19 Hz)。HRMS (ESI) 精确质量计算值 C35H24F5N4[M+H]+:595.19211,实验值595.19197。
5-五氟苯基-15-(2,4,6-三甲氧基苯基)卟啉 (4c) 紫色固体,0.0522 g产率9.8%。1H NMR (400 MHz,CDCl3) δ 10.27 (s,2H,meso-H),9.43 (d,2H,J=4.4 Hz,pyrrole-Hβ),9.34 (d,2H,J=4.4 Hz,pyrrole-Hβ),9.02 (d,2H,J=4.4 Hz,pyrrole-Hβ),8.94 (d,2H,J=4.4 Hz,pyrrole-Hβ),6.63 (s,2H,Phenyl-H),4.14 (s,3H,PhOCH3),3.53 (s,6H,2PhOCH3),-3.11 (s,2H,-NH-)。19F NMR (376 MHz,CDCl3) δ -136.70 (dd,2F,J=9.78,25.57 Hz),-153.15 (t,1F,J=21.81 Hz),-162.36 (td,2F,J=8.65,24.44 Hz)。HRMS (ESI) 精确质量计算值 C35H24F5N4O3[M+H]+:643.176 86,实验值643.176 94。
5-(2,6-二甲氧基苯基)-15-(五氟苯基)卟啉 (4 d) 紫色固体,0.2052 g产率40.4%。1H NMR (400 MHz,CDCl3) δ 10.28 (s,2H,meso-H),9.43 (d,2H,J=4.4 Hz,pyrrole-Hβ),9.34 (d,2H,J=4.4 Hz,pyrrole-Hβ),9.02 (d,2H,J=4.4 Hz,pyrrole-Hβ),8.95 (d,2H,J=4.4 Hz,pyrrole-Hβ),7.79 (t,1H,J=8.4 Hz,Phenyl-H),7.06 (d,2H,J=8.8 Hz,Phenyl-H),3.55 (s,6H,2PhOCH3),-3.10 (s,2H,-NH-)。19F NMR (376 MHz,CDCl3) δ -136.69 (dd,2F,J=9.40,25.19 Hz),-153.11 (t,1F,J=21.43 Hz),-162.33 (td,2F,J=9.40,25.19 Hz)。HRMS (ESI) 精确质量计算值C34H22F5N4O2[M+H]+:613.16629,实验值613.16647。
5-(2,6-二氯苯基)-15-(五氟苯基)卟啉 (4e) 紫色固体,0.0761 g产率14.8%。1H NMR (400 MHz,CDCl3) δ 10.34 (s,2H,meso-H),9.45 (d,2H,J=4.4 Hz,pyrrole-Hβ),9.41 (d,2H,J=4.4 Hz,pyrrole-Hβ),8.97 (d,2H,J=4.0 Hz,pyrrole-Hβ),8.91 (d,2H,J=4.4 Hz,pyrrole-Hβ),7.85 (d,2H,J=7.6 Hz,Phenyl-H),7.76 (t,1H,J=8.0 Hz,Phenyl-H),-3.15 (s,2H,-NH-)。19F NMR (376 MHz,CDCl3) δ -136.68 (dd,2F,J=8.65,25.94 Hz),-152.66 (t,1F,J=22.18 Hz),-162.08 (td,2F,J=7.90,24.06 Hz)。HRMS (ESI) 精确质量计算值 C32H16F5N4Cl2[M+H]+:621.067 22,实验值621.066 98。
5-(3,5-二叔丁基苯基)-15-(五氟苯基)卟啉 (4f) 紫色固体,0.0777 g产率14.1%。1H NMR (400 MHz,CDCl3) δ 10.34 (s,2H,meso-H),9.46 (s,2H,pyrrole-Hβ),9.41 (s,2H,pyrrole-Hβ),9.17 (s,2H,pyrrole-Hβ),8.99 (s,2H,pyrrole-Hβ),8.15 (s,2H,Phenyl-H),7.89 (s,1H,Phenyl-H),-3.04 (s,2H,-NH-)。19F NMR (376 MHz,CDCl3) δ -136.63 (dd,2F,J=7.52,23.69 Hz),-152.86 (t,1F,J=21.06 Hz),-162.14 (td,2F,J=8.27,23.69 Hz)。DEPT-135 (100 MHz,CDCl3) δ 132.78,131.79,131.41,130.01,128.62,121.18,105.68,31.56。HRMS (ESI) 精确质量计算值 C40H34F5N4[M+H]+:665.270 36,实验值665.271 83。
2 结果与讨论
2.1 反应条件优化
首先,以二吡咯甲烷和五氟苯甲醛的缩合反应为模型反应,进行反应条件优化,筛选结果见表1。
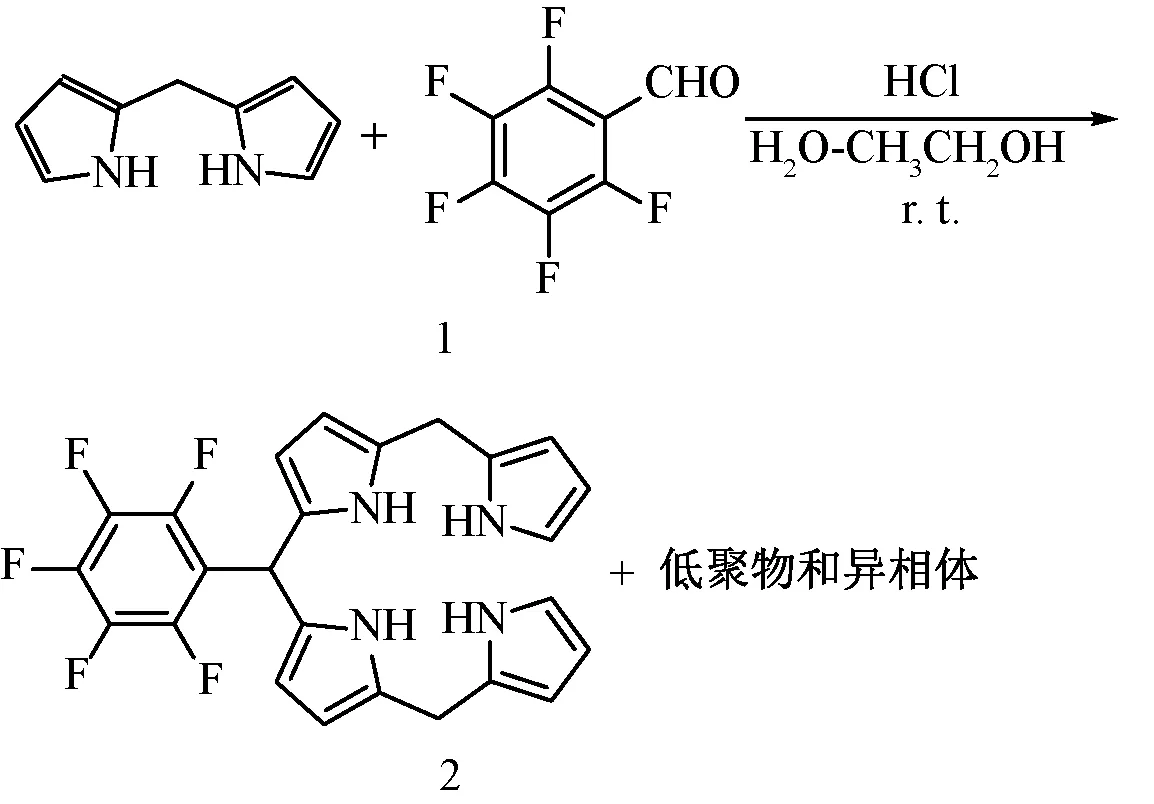
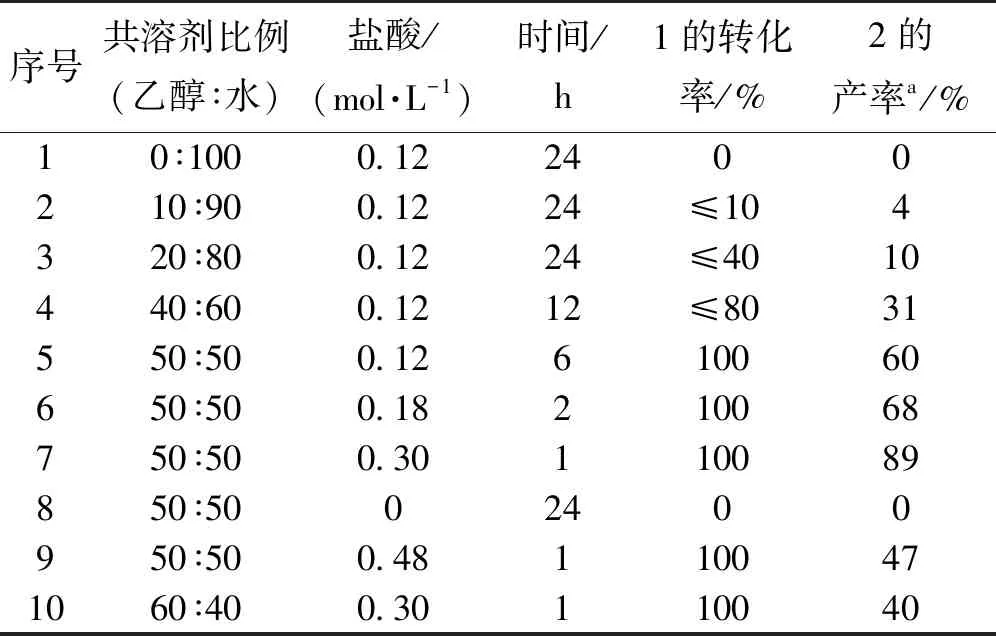
表1 水相中不同条件下五氟苯甲醛与二吡咯甲烷的缩合反应
众所周知,反应的核心是溶解度,基于二吡咯甲烷1本身在水中的溶解度有限,要使二吡咯甲烷与五氟苯甲醛的缩合反应能够在水相中顺利进行,就必须找到一种与水互溶的溶剂,在合适的比例下维持二吡咯甲烷在此共溶剂中有相当的溶解度,而目标产物10-五氟苯基四吡咯烷在此共溶剂中溶解度很小,生成的10-五氟苯基四吡咯烷在此共溶剂中析出,从而使反应不断向右进行。基于前期实验选择较经济的乙醇作为共溶剂,0.12 mol/L盐酸浓度作为初始实验的催化剂浓度,首先调查在纯水中实验和TLC监测反应的情况,结果与预期的结果一致,反应24 h后始终没有检测到四吡咯烷的生成(表1中序号 1,紫外灯下二吡咯甲烷在薄板上呈现黄色荧光斑点,四吡咯烷为暗褐色荧光斑点),因此,乙醇作为共溶剂以不同的体积比例加入反应体系中,乙醇的添加比例范围从10%~60%,当乙醇-水共溶剂比例过小时,反应24 h后原料二吡咯甲烷大部分没有消耗掉,TLC可以监测到10-五氟苯基四吡咯烷的生成(表1中序号 2、3),当提高乙醇与水的共溶剂比例达到40:60时,反应经历12 h,TLC监测有80%的二吡咯甲烷消耗掉了,相应的四吡咯烷的产率仅仅达到31%左右(表1中序号 4),受此鼓舞,当提高乙醇与水的共溶剂比例达到50:50时,二吡咯甲烷能够在6 h内完全消耗掉,并且10-五氟苯基四吡咯烷的柱层析分离收率达到60%左右(表1中序号 5),当增加盐酸催化剂的浓度时,发现二吡咯甲烷很快消耗完(表1中序号6),四吡咯烷的产率也有相应提高,当把盐酸浓度提高到0.3 mol/L,二吡咯甲烷在1 h内就可以消耗掉,并且10-五氟苯基四吡咯烷的收率达到89%(表1中序号7),控制实验表明,反应体系不加盐酸,此缩合反应不发生(表1中序号8),继续增加盐酸浓度到0.48 mol/L,并没有得到更好的结果,10-五氟苯基四吡咯烷产物的分离收率开始有明显下降(表1中序号9),更高浓度盐酸加入可能导致其它寡聚物的生成。从而四吡咯烷分离收率有明显下降。此外,当共溶剂比例增加时,10-五氟苯基四吡咯烷的分离收率反而下降明显(表1中序号10)。在此共溶剂条件下,10-五氟苯基四吡咯也可能有一定的溶解度,出现竞争反应,导致10-五氟苯基四吡咯烷的分离收率明显下降。最优的的反应条件如下:50%的乙醇水溶液作为共溶剂,盐酸的浓度为0.30 mol/L,相应的10-五氟苯基四吡咯烷柱层析分离收率达到87%。
接着,在水相中合成卟啉就成了着手准备的工作,以10-五氟苯基四吡咯烷和对甲基苯甲醛的缩合反应为模型反应,筛选最佳反应条件,结果见表2。
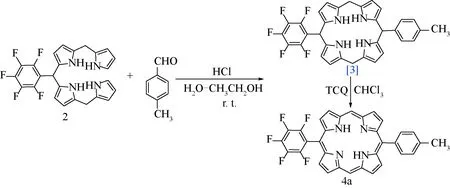
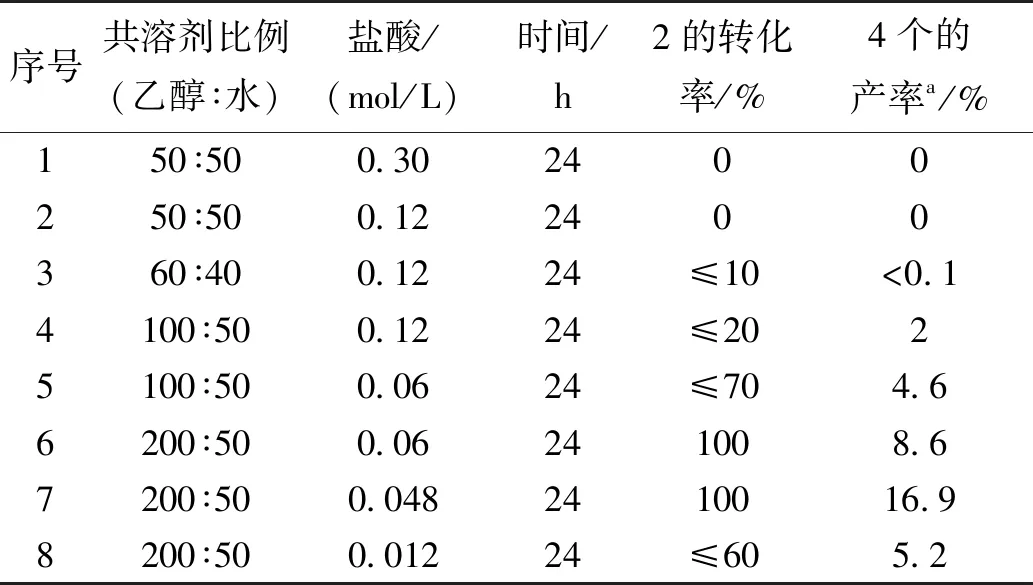
表2 水相中不同条件下对甲基苯甲醛与10-五氟苯基四吡咯烷的缩合反应
同样在实验前,经过分析可知,10-五氟苯基四吡咯烷在1∶1的乙醇水共溶剂中的溶解度有限,要使10-五氟苯基四吡咯烷与芳醛在水相中能进行缩合反应,就必须提高共溶剂比例,使反应原料尽可能在均相中反应,这样缩合反应就会进行,最终的卟啉原就会从共溶剂中析出而使反应不断地向右进行,正如可预计的实验结果,较低的共溶剂比例(表2中序号 1至5),反应24 h,几乎得不到想要的目标产物(肉眼下卟啉原呈现一个清晰粉红色荧光斑点,卟啉为深红色荧光斑点),经过对反应条件优化,当把共溶剂比例提高到4∶1时,盐酸浓度降低至0.048 mol/L,4a获得最好的收率16.9%(表2中序号6),再继续降低盐酸的浓度,反而原料10-五氟苯基四吡咯烷在给定的24 h内消耗不完(表2中序号7)。最终的水相合成卟啉原的优化条件如下:4∶1的乙醇水作为共溶剂,盐酸的浓度为0.048 mol/L,室温下避光反应24 h。
在相应优化条件下,其它位阻芳醛给予测试,2,4,6-三甲基苯甲醛,2,4,6-三甲氧基苯甲醛,2,6-二甲氧基苯甲醛,2,6-二氯苯甲醛和3,5-二叔丁基苯甲醛都可以以可观的产率获得相应的卟啉。
2.2 反应机理推测
这个反应机理如图2所示,五氟苯甲醛羰基氧在盐酸催化下质子化,有利于二吡咯甲烷α碳进攻生成傅克烷基化中间体再质子化失水生成中间体A,另一分子的二吡咯甲烷α碳进攻中间体A,失质子生成10-五氟苯基四吡咯烷2。同样在生成的卟啉原的过程中,芳醛醛羰基氧在盐酸催化下质子化,有利于10-五氟苯基四吡咯烷2的吡咯α碳进攻生成傅克烷基化中间体再质子化失水生成中间体B,10-五氟苯基四吡咯烷2的另一吡咯α碳进攻中间体B,失质子生成5-五氟苯基-15-芳基取代卟啉原。

图2 假定的机理
3 结论
(1)本文通过实验证实了在80%的乙醇水溶液中,0.048 mol/L盐酸催化下,室温下5-(五氟苯基)四吡咯甲烷可以与另一分子的芳醛发生缩合反应,生成卟啉原继而氧化生成Trans-AB卟啉;通过上述方法成功合成了6种未见文献报道的Trans-AB位阻卟啉,并且通过核磁氢谱、核磁碳谱、核磁氟谱和高分辨质谱确证了相应化合物结构。
(2)上述催化体系具有反应条件温和、反应简单、底物适用范围广、原料廉价易得等优点,在卟啉的仿生催化剂设计及合成方面具有重要的应用前景和实用价值。
