新生儿甲基丙二酸血症筛查及随访分析
2022-03-04张振季婵婵董丽萍李庆波
张振 季婵婵 董丽萍 李庆波



[摘要] 目的 了解淄博市新生兒甲基丙二酸血症(MMA)的总体发病率,分析MMA合并型临床特征、基因突变特点及预后。方法 通过串联质谱技术筛查本市2013 年10 月至2019年7月出生且监护人自愿选择筛查的所有新生儿样本,对诊断为MMA的新生儿进行基因检测。 结果 筛查185 239例新生儿中共确诊44例,单纯型5例,合并型39例; 39例合并型治疗随访时间9 d~6.5岁不等,新生儿期死亡3 例,27例无症状,9例有症状,表现为不同程度的眼球震颤、癫痫、脑积水和智能发育落后等;血液同型半胱氨酸浓度与尿中MMA浓度Spearman相关分析P=0.474。MMACHC基因检测突变位点检出率100.00%,检出16种突变类型,携带c.482G>A基因的患儿预后好;死亡病例分别携带c.609G>A/c.80A>G;c.80A>G纯合突变;c.277-3_303del/c.658_660del AAG。 结论 淄博市甲基丙二酸血症发病率为1/4210,合并型甲基丙二酸血症基因型与表型相关,同型半胱氨酸浓度升高与预后相关,与尿中MMA浓度无密切相关。早诊断、早期规范治疗预后好。
[关键词] 新生儿疾病筛查;甲基丙二酸血症;同型半胱氨酸;尿中MMA浓度;随访
[中图分类号] R722.1 [文献标识码] B [文章编号] 1673-9701(2022)02-0084-04
Screening and follow-up analysis of neonatal methylmalonic acidemia
ZHANG Zhen1 JI Chanchan1 DONG Liping1 LI Qingbo2
1.Neonatal Screening Center, Zibo Maternal and Child Health Hospital in Shandong Province, Zibo 255000, China; 2.Zhejiang Biosan Biochemical Technologies Co., Ltd. Hangzhou 310000, China
[Abstract] Objective To investigate the overall incidence of neonatal methylmalonic academia (MMA) in Zibo city, and to analyze the clinical characteristics, gene mutation characteristics and prognosis of combined MMA. Methods Tandem mass spectrometry was used to screen all newborns born from October 2013 to July 2019 in the city whose guardians voluntarily selected screening. Genetic testing was performed on newborns diagnosed with MMA. Results Among the 185 239 newborns screened, there were 44 newborns diagnosed with MMA, including 5 with simple MMA and 39 with combined MMA. In the 39 newborns with combined MMA, the follow-up time ranged from 9 days to 6.5 years. There were 3 newborns died in neonatal period, 27 asymptomatic newborns, and 9 symptomatic newborns, manifested as varying degrees of nystagmus, epilepsy, hydrocephalus, and mental retardation. Spearman correlation analysis of blood homocysteine concentration and urine MMA concentration showed P=0.474. The detection rate of mutation sites in MMACHC gene detection was 100.00%, and 16 mutation types were detected. Children with c.482G>A gene had a good prognosis. Dead newborns carried c.609G>A/c.80A>G, c.80A>G homozygous mutation, and c.277-3_303del/c.658_660del AAG. Conclusion The incidence of MMA in Zibo city is 1/4210. The genotype of combined MMA is related to the phenotype. The increase of homocysteine concentration is related to the prognosis, and it is not closely related to the urine MMA concentration. Early diagnosis and early standard treatment promise good prognosis.
[Key words] Neonatal disease screening; Methylmalonic acidemia; Homocysteine; Urine MMA concentration; Follow-up
甲基丙二酸血症(methylmalonic academia,MMA)是一种常染色体隐性遗传病,由于甲基丙二酰辅酶A变位酶自身缺陷或其辅酶钴胺素代谢缺陷导致患者体内多种氨基酸代谢障碍,引起甲基丙二酸、3-羟基丙酸及甲基枸橼酸等代谢物异常蓄积,影响同型半胱氨酸代谢[1]。根据MMA患者血同型半胱氨酸水平正常或增高这一生化差异可将其分为单纯型MMA和MMA合并同型半胱氨酸血症两种类型[2]。串联质谱筛查技术在新生儿疾病筛查中的广泛应用,部分甲基丙二酸血症患者可在新生儿期被检出,大大降低了患儿病残率,有效地提高出生人口素质。淄博市在2013年10月开展利用串联质谱技术进行MMA的筛查,覆盖率约占出生人口的60%以上。本研究旨在了解淄博市新生儿MMA的总体发病率,分析MMA合并型临床特征、基因突变特点及预后,现报道如下。
1 资料与方法
1.1 一般资料
选取2013年10月至2019年7月在淄博市出生且在監护人知情同意下进行筛查的样本185 239例。
1.2 方法
1.2.1 筛查样本的采集 新生儿充分哺乳6次,生后3~7 d,最迟不超过生后20 d,采集足底血滴于专用滤纸片,室温下自然晾干,于4℃保存,1周内递送(冷链运输)淄博市妇幼保健院中心实验室进行检测。
1.2.2 实验室检测 应用Waters2777c自动进样器,Waters 1525 Binary HPLC Pump(1525高效液相色谱泵)和Waters-TQ Detector串联质谱仪检测,PE公司提供的试剂盒,使用非衍生法检测新生儿体内各种氨基酸和酰基肉碱浓度及比值。步骤如下:①整理标本,使用洁净的U型截底微孔板按照布板进行干血斑打孔。取出内标储备液,室温放置2 min,振荡孵育器打开预热。使用内标储备液按照1∶110稀释比稀释配制内标工作液,混匀。②每孔加入100 μl内标工作液,使用粘性封套覆盖微孔板,在振荡孵育器中振荡45 min。③振荡结束后,稍放置冷却,每孔转移75 μl至V型板中,用铝膜密封。即可上机检测。
1.2.3 基因检测 经家长知情同意,取患儿及父母外周血标本进行二代高通量测序,检测到基因突变并进行父母Sanger 测序验证,检测到单个变异位点者加用q-PCR 方法检测。本研究经医院医学伦理委员会批准并全程监督。
1.2.4 召回标准 串联质谱(MS/MS)检测丙酰基肉碱(C3)、丙酰基肉碱/乙酰基肉碱(C3/C2)升高[1],2013年10月至2016年6月参考范围C3(0.42~4.90),C3/C2(0.03~0.20);2016年7月至2019年7月C3(0.39~4.90)、C3/C2(0.03~0.18),单位μmol/L。
1.2.5 诊断标准 单纯型符合下述①+②+③+④合并或不合并⑦即可诊断;合并型符合下述①+②+③+⑤合并或不合并⑥即可诊断。①MS/MS:C3/C2 升高,合并或不合并C3升高。②尿有机酸分析:甲基丙二酸升高。③维生素B12、叶酸正常。④同型半胱氨酸正常。⑤同型半胱氨酸升高。⑥MMACHC基因检测杂合突变或纯合突变。⑦MUT基因检测杂合突变或纯合突变。
1.2.6 治疗与随访 合并型给予左卡尼汀口服液[东北制药,国药准字H19990372,规格:1 g/(10 ml·支)]50~100 mg/(kg·d)、维生素B12注射液[容生制药,国药准字H41020633,500 μg/(ml·支)]1 mg/d,2~5次/周、亚叶酸钙片(广东岭南制药国药准字H20040396,以亚叶酸计15 mg/片)3.75 mg/d,1~2次/d、甜菜碱(保健品无国药准字号)0.25 g/(kg·d)及其他对症治疗;单纯型左卡尼汀和特殊奶粉及对症治疗。早期维生素B12注射液1 mg/d肌肉注射 3~5 d复查,病情稳定后改为1 mg/次,2~5次/周,1~3个月复查。采用门诊和电话随访方式,随访时间9 d~6.5岁。
1.3 统计学方法
采用SPSS 22.0统计学软件进行数据分析,采用两相关样本的Wilcoxon符号秩和检验,比较在初筛和复筛样本中甲基丙二酸确诊患儿氨基酸及酰基肉碱水平,并利用中位数法进行统计描述;采用Spearman相关分析血液中同型半胱氨酸和尿中甲基丙二酸浓度值关系;采用百分位数统计合并型MMA基因突变位点频率结果,P<0.05为差异有统计学意义。
2 结果
2.1 确诊情况及发病率
2013年10月至2016年6月筛查75 932例召回确诊 32 例,确诊MMA 19例,VitB12缺乏4例;2016年7月至2019年7月筛查109 307例,召回确诊 80例,确诊MMA 25例,VitB12缺乏20例。总体召回确诊人数112例,阳性预测值38.39%,共确诊MMA 44例,其中合并型39例,单纯型5例,总体发病率1/4210。随访合并型39例,其中男23例,女16例。诊断均在新生儿期完成,后经基因验证诊断。治疗开始时间9~28 d。
2.2 合并型患儿初筛与复筛各项酰基肉碱及氨基酸水平比较
初筛和复筛C3/C2比较,C3/C2复筛值明显高于初筛值,差异有统计学意义(P<0.001)。见表1。
2.3 VitB12负荷试验前HCY和尿中MMA浓度Spearman相关分析
VitB12负荷试验前HCY浓度与尿中MMA浓度Spearman相关分析,相关系数0.120,差异无统计学意义(P=0.474>0.05)。见表2。
2.4 随访结果
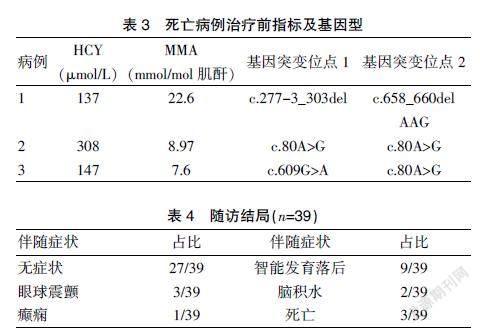
39例合并型患者中早发型3例,召回确诊时已发病,负荷试验未完成于新生儿期死亡,HCY浓度明显升高,但尿中MMA只有轻度升高。见表3。27例无症状,治疗后尿中MMA和MSMS正常,HCY<30 μmol/L;9例有症状,表现为不同程度的眼球震颤、癫痫、脑积水和智能发育落后等,随访中MSMS正常,尿中MMA正常或轻度升高,HCY>50 μmol/L。携带c.482G>A复合杂合突变基因患者13例,VitB12负荷试验后各项指标恢复正常快,预后好。见表4。
2.5 合并型MMA患儿基因检测结果
39例患儿均检测出2个基因变异,共检测出MMACHC基因16种变异,其中c .609G>A变异频率最高,其次是c.482G>A是当地人群的热点突变。检出1例罕见的1号外显子缺失患儿。见表5。
3讨论
MMA是较常见的有机酸尿症,发病率在不同国家有很大差异,美国发病率为1/2.9万,加拿大发病率为1/6.1万[3]。上海新华医院筛查76万例新生儿发病率为1/3.33万,浙江省儿童医院筛查近13万例,发病率为1/6.66万[4],河南省发病率为1/6032[5],山东聊城发病率为1/5578[6],山东潍坊地区发病率为1/6498[7],本市发病率为1/4210,表明该病有明显的地区差异,山东、河南是该病的高发地区。
甲基丙二酸是异亮氨酸、甲硫氨酸、苏氨酸、缬氨酸、胆固醇和奇数链脂肪酸分解、代谢途径中甲基丙二酰辅酶A的代谢产物,正常情况下在甲基丙二酰辅酶A变位酶(methylmalonyl-CoA mutase,MCM)及其辅酶腺苷钴胺素(AdoCbl,由VitB12代谢而来)的作用下转化并生成琥珀酸,参与三羧酸循环。当机体内甲基丙二酰辅酶A变位酶出现自身缺陷或维生素B12代谢缺陷,就会导致甲基丙二酰辅酶A代谢途径受阻,串联质谱中表现为丙酰基肉碱(C3)升高,乙酰基肉碱(C2)生成相对减少,可表现为C3/C2升高,所以MSMS检测主要指标C3和C3/C2升高[4],当其辅酶腺苷钴胺素代謝障碍时会生成蛋氨酸减少,合并同型半胱氨酸升高,所以蛋氨酸降低可作为MMA诊断和类型鉴别的一个参考指标[8]。尿中有机酸分析可见甲基丙二酸、甲基枸橼酸、3-羟基丙酸等代谢物蓄积。合并型患儿初筛与复筛各项酰基肉碱及氨基酸水平比较发现,初筛和复筛C3/C2 水平比较,差异有统计学意义(P<0.001),随着采血年龄的增加,病情加重,C3/C2浓度值水平不断升高,C3/C2浓度值水平不断升高,说明C3/C2比C3更有价值[9]。
MMA患儿可发病于任何年龄,临床表现缺乏特异性,可表现为多系统器官损伤,其中脑神经损伤最为突出,若不及时诊治,患者致死、致残率较高[10]。根据发病年龄可将MMA分为早发型和迟发型,早发型多在1岁内起病,多表现为喂养困难、呕吐、抽搐、酸中毒等症状,大部分患者留有严重后遗症,迟发型少见,多在4岁后出现智力、运动能力倒退、记忆力下降、认知障碍等[11]。本研究对象均来源于新生儿筛查群体,但由于血斑样本递送、分析和报告时间的安排,有3例患儿为早发型,在召回确诊时已发病,负荷试验未完成即夭折,HCY浓度明显升高,但尿中MMA极低,甚至接近正常,其尿中MMA低可能与病情重、喂养困难、摄入氨基酸少及肾脏代谢能力下降等因素有关;研究对象中部分迟发型患者治疗前尿中MMA也常出现极低的情况,所以治疗前尿中MMA浓度高低不能准确评估预后。同型半胱氨酸为含巯基氨基酸,是胱硫醚和蛋氨酸转硫化和甲基化代谢旁路中形成的中间体,同型半胱氨酸蓄积可导致血管内皮损伤和血液系统异常如血栓性血小板减少、溶血尿毒综合征[12]、骨髓抑制[13],同型半胱氨酸浓度持续升高会导致患儿以神经系统症状、贫血、肺动脉高压为首发症状就诊。少数研究对象诊断初期伴有轻度喂养困难,及时给予维生素B12负荷试验后症状消失,随访中仍可观察到逐渐出现以眼球震颤为主要表现的神经系统损害,MSMS基本正常,尿中MMA正常或轻度升高,只有同型半胱氨酸浓度持续升高,提示同型半胱氨酸浓度升高与预后相关,统计学分析同型半胱氨酸浓度与尿液中MMA无密切相关性(P>0.05),发病越早,预后越差。
MMA是常染色体隐性遗传病,CblC亚型是其中较常见的类型,基因型为MMACHC型,该型主要累及神经系统[9]。MMACHC基因位于1p34.1,包含5个外显子,其中1~4号外显子为编码区,5号外显子为非编码区,共编码282个氨基酸,共发现有80余种突变,且突变多集中于3、4号外显子[14-15]。研究对象39例患者均为MMACHC型,共检测出16种突变类型,占比前三位分别为c.609G>A(30.77%)、c.482G>A(16.67%)、c.658_660del AAG(12.82%),与文献报道[16-17]前三位的突变位点分别为c.609G>A、c.80A>G和c.482G>A不完全相同。有文献阐述[18-20],c.80A>G和c.658_660del AAG、c.80A>G纯合突变及c.80A>G/c.609G>A杂合突变,均为早发型临床表现重预后差,研究中的死亡病例携带上述的突变位点与文献中观点相同。c.482G>A基因突变使cblC蛋白第161位氨基酸由精氨酸变为谷氨酰胺,对蛋白质功能的影响较小,因此发病较晚,国内外研究发现该位点突变与晚发型 MMA有一定相关性,且临床表现较轻[18,21-23],但仍有较高发病风险[19],随访病例中13例携带c.482G>A 基因突变,他们的负荷试验显效,随访中虽无临床症状,但仍可观察到同型半胱氨酸随着年龄增加逐渐升高趋势,进而可能造成进行性神经系统损害,出现临床症状,所以携带c.482G>A基因的患儿仍需规律用药,降低该突变的早发风险。
综上所述,淄博市甲基丙二酸血症发病率为1/4210;合并型甲基丙二酸血症基因型与表型相关,同型半胱氨酸浓度升高与预后相关,与尿中MMA浓度无密切相关;早诊断、早期规范治疗预后好。
[参考文献]
[1] 鄂慧姝,韩连书,叶军,等.甲基丙二酸血症MMACHC基因c.482G>A突变患者临床资料及随访分析[J].中华内分泌代谢杂志,2019,35(7):581-585.
[2] Fowler B,Leonard JV,Baum gartner MR.Causes of and diagnostic approach to methylm a lonic acidurias[J]. J Inherit Metab Dis,2008,31(3):350-360.
[3] Ma X,Zhang Y,Yang Y,et al.Epilepsy in children with methylmalonic academia:Electroclinical features and prognosis[J].Brain Dev,2011,33(9):790-795.
[4] 顾学范.临床遗传代谢病[M].北京:人民卫生出版社,2015.
[5] 赵德华,朱昕赟,李晓乐,等.河南省349858例新生儿甲基丙二酸血症(MMA)的筛查结果分析[J].中国优生与遗传杂志,2016,24(8):86-87,90.
[6] 刘明芳.聊城地区应用串联质谱技术筛查新生儿甲基丙二酸血症分析[J].中国优生与遗传杂志,2018,26(2):29-30.
[7] 裴薇,鹿相花,田维兵,等.潍坊地区新生儿遗传代谢病串联质谱筛查结果分析[J].中国中西医结合儿科学,2018,10(5):452-455.
[8] Weisfeld-Adams JD,Morrissey MA,Kirrnse BM,et al.New bom screening and early biochemical follow-up in combined methylm aonic aciduria and homocystinuria,Cbl-Ctype,and utility of methionine as a secondary screening analyte[J]. Mol Genet Metab,2010,99(2):116-123.
[9] 黄倬,韩连书,叶军,等.甲基丙二酸血症患者143例资料分析[J].中华内分泌代谢杂志,2014,30(6):490-494.
[10] Weisfeld-Adams JD,McCourt EA,Diaz GA,et al.Ocular disease in the cobalamin C defect:A review of the literature and a suggested framework for clinical surveillance[J].Mol Genet Metab,2015,114(4):537-546.
[11] Hu S,Mei S,Liu N,et al.Molecular genetic characterization of cblC defects in 126 pedigrees and prenatal genetic diagnosis of pedigrees with combined methylmalonic aciduria and comocystinuria[J].BMC MedGenet,2018,19(1):154.
[12] Menni F,Testa S,Guez S,et al. Neonatal atypical hemolytic uremic syndrome due to methylmalonic aciduria and homocystinuria[J].Pediatr Nephrol,2012,27(8):1401-1405.
[13] Watkins D,Rosenblatt DS.Inborn errors of cobalamin absorption and metabolism[J].Am J Med Genet C,2011, 157C(1):33-44.
[14] Kang LL,Li YP,Shen M,et al.A study on a cohort of 301 Chinese patients with isolated methylmalonic academia[J].J Inherit Metab Dis,2019,43(3):409-423.
[15] 劉玉鹏,杨艳玲.甲基丙二酸尿症cblC型合并同型半胱氨酸血症的临床与实验室研究进展[J].中华儿科杂志,2013,51(4):313-316.
[16] Wang C,Li D,Cai FY,et al.Mutation spectrum of MMACHC in Chinese pediatric patients with cobalamin C disease:A case series and literature review[J].European Journal of Medical Genetics,2019,62(10):103 713.
[17] 刘畅,孟翠萍,房振楠,等.菏泽地区新生儿甲基丙二酸血症的筛查及基因突变分析[J].中国儿童保健,2021, 29(4):372-376.
[18] Liu MY,Yang YL,Chang YC,et al.Mutation spectrum of MMACHC in Chinese patients with combined methylmalonic aciduria and homocystinuria[J]. J Hum Genet,2010,55(9):621-626.
[19] Wang F,Han LH,Yang YL,et al. Clinical,biochemical,and molecular analysis of combined methylmalonic acidemia and hyperhomocysteinemia(cblC type)in China[J].J Inherit Metab Dis,2010,33(Suppl 3):S435-S442.
[20] Martina Huemer,Sabine Seholl-Sargi,et al.Three new cases oflate-onset cblC defect and review of the literature illustrating when to consider inbom errors ofmetabolism beyond infancy[J].Orphanet J Rare Diseases,2014,9:161.
[21] 赵培伟,蔡晓楠,吴革菲,等.甲基丙二酸血症3例报告并文献复习[J].临床儿科杂志,2016,34(12):894-897.
[22] Lerner-Ellis JP,Anastasio N,Liu J,et al.Spectrum of mutations in MMACHC,allelic expression,and evidence for genotype-phenotype correlations[J].Hum Mutat,2009, 30(7):1072-1081.
[23] Tsai AC,Morel CF,Scharer G,et al.Late-onset combined homocystinuria and methylmalonic aciduria(cblC) and neuropsychiatric disturbance[J].Am J Med Genet A,2007, 143A(20):2430-2434.
(收稿日期:2021-06-16)
