高速逆流色谱结合半制备液相色谱制备甘青青兰中3种活性成分
2022-03-02周加本热增才旦热毛先利毛才让
周加本 热增才旦 热毛先 利毛才让,3*
(1. 青海师范大学生命科学学院,西宁 810008;2. 青海民族大学药学院,西宁 810007;3. 青海师范大学民族师范学院,西宁 810008)
高速逆流色谱(HSCCC)是一种高效分离天然药物活性成分的新型技术,具有制备量多、操作简便、分离快速、纯度高、无需载体支撑以及两相溶剂的选择和组合多样灵活等特点,因此,该技术被广泛应用天然活性物质的分离制备领域。高速逆流色谱与制备液相色谱(Pre-HPLC)技术相结合进行分离纯化,可快速高效地分离制备单体化合物,对藏药对照品的制备方法研究具有重要意义。
甘青青兰(Maxim.),又名唐古特青兰,为唇形科(Lamiaceae)青兰属(Linn)植物,是藏医药临床常用药材,具有止咳化痰、和胃舒肝、清热解毒的功效。甘青青兰中有酚酸类、黄酮及黄酮苷类等成分,并有良好的抗氧化、抗病毒、抗心脑缺氧、抗炎、抑菌等药理活性,其中绿原酸、胡麻甙-6″-乙酯和迷迭香酸是甘青青兰主要活性成分,且具有抗氧化、保肝和治疗心脑缺氧的药理作用。目前,从甘青青兰中分离制备绿原酸、胡麻甙-6″-乙酯和迷迭香酸方法仅限于传统技术,如硅胶柱色谱、凝胶柱色谱、高效液相色谱等,这些方法存在实验周期长、成本高、制备量小等缺点。本研究建立了高速逆流色谱和制备液相色谱相结合,快速、高效分离制备甘青青兰中绿原酸、胡麻甙-6″-乙酯和迷迭香酸(见图1)的方法。该方法分离时间短,产物纯度高,制备量大,对进一步研究甘青青兰及其复方的质量控制提供了参考,为甘青青兰中绿原酸、胡麻甙-6″-乙酯和迷迭香酸的药理及其作用机制研究提供了基础。

图1 目标化合物结构式Fig.1 Chemical structure of target compound
1 仪器与材料
TBE-1000 高速逆流色谱仪(上海同田生化技术有限公司),TBP-2H02 泵(上海同田生化技术有限公司),UV-300N 全波长紫外检测器(上海三为科学仪器有限公司),N2010 记录仪(浙江大学智达信息工程有限公司);DAC-50 制备型液相色谱仪(江苏汉邦科技有限公司,配有NP-7000 泵和NU-3000 UV/VIS 检测器),V220B 色谱记录仪(北京晨悦浩祥公司);LC-3100 皖仪高效液相色谱仪(安徽皖仪科技股份有限公司,配有P3200Q 四元泵和DAD-3100 检测器);Varian INOVA-600 核磁共振波谱仪(美国Varian公司)。
高速逆流色谱和制备液相用试剂均为分析纯(济南化学试剂厂),乙腈为色谱纯(山东禹王公司),水为超纯水。
甘青青兰采购于青海省八一路药材市场,由青海大学杨仕兵教授鉴定为青兰属植物甘青青兰的干燥地上部分。
2 试验方法
2.1 甘青青兰粗提物的制备
将干燥甘青青兰(DTM)药材5 kg 制成粉末后,加15 倍量75%乙醇,在75 ℃下回流提取3 次,每次2 h,合并提取液,减压浓缩,将浓缩液移入分液漏斗中,依次用石油醚(A)、乙酸乙酯(B)、正丁醇(C)反复萃取,收集各部位萃取液,减压回收溶剂,得乙酸乙酯部位浸膏172.9 g,冷藏供分离。
2.2 高效液相色谱(HPLC)条件
色 谱 柱:Welch Ultimate MB-C(4.6 mm ×250.0 mm,5 µm);流动相:乙腈(A)-0.1%醋酸溶液(B);梯度洗脱(0~30 min,10%~48% A;30.0~30.1 min,48%~10%A;30.1~35.0min,10%A);流速为1 mL·min;柱温为30 ℃;多波长检测(200~400 nm);进样量为15µL。
2.3 半制备型高效液相(SP-HPLC)分离富集
称取1.00 g 甘青青兰乙酸乙酯萃取物,加入10 mL 50%甲醇溶解,离心取5 mL 上清液,上样于DAC-50 半制备型高效液相,进行梯度洗脱,根据色谱峰分别收集13.12~15.05 min(DTM-B-1)和17.54~25.00 min(DTM-B-2)时的流出液,减压浓缩至干。取少量干粉按“2.2”项下方法进行HPLC检测。
色谱条件:Megres C(20 mm×250 mm,10µm)柱;流动相为甲醇(A)-0.4%冰醋酸溶液(B);洗脱程序为(0~6 min,10% A;7~30 min,10%~50% A;31~45 min,50%~95% A);流速为15 mL·min;柱温为30 ℃;检测波长为340、360 nm。
2.4 HSCCC分离
2.4.1 分配系数的测定
称取10 mg“2.3”项分离得DTM-B-2 加入两相溶剂体系中,涡旋振荡使样品充分溶解于两相中,静置分层,分别取上、下相按“2.2”项下方法进行检测,目标物在上、下中的峰面积比值为分配系数(值)。
2.4.2 HSCCC分离条件
将两相溶剂体系中的下相以50 mL·min的流速泵入逆流色谱仪的螺旋管中,待充满螺旋管后,开启仪器,调节主机至FWD 模式进行旋转,转速稳定在850 r·min后,以5 mL·min流速泵入上相,当有流动相从出口稳定流出,即螺旋管中的两相已达到平衡后,将样品溶液由进样阀注入逆流色谱仪,流速调至2.5 mL·min,开启检测器和记录仪,检测波长为340 nm。
2.5 Pre-HPLC 纯化条件
色谱条件:Megres C(20 mm×250 mm,10µm)柱;流动相为甲醇(A)-0.4%冰醋酸(B);柱温为25 ℃;检测波长为340、360 nm;流速为15 mL·min;进样量为1.5 mL。
3 结果与分析
3.1 SP-HPLC分离富集
HPLC 检测结果显示,DTM-B-1 和DTM-B-2 中目标化合物含量较高(见图2),故重复进行“2.3”项中对DTM-B-1 和DTM-B-2 的收集,减压浓缩,冷冻干燥,获得1.50g DTM-B-1和7.44 g DTM-B-2,待分离。

图2 乙酸乙酯部位(A)及其半制备型液相分离(B),DTM-B-1(C),DTM-B-2(D)的色谱图Fig.2 Chromatograms of ethyl acetate(A)and Semi-pre⁃parative separation(B),DTM-B-1(C),DTM-B-2(D)
3.2 化合物Ⅰ的纯化
经“3.1”项分离可得化合物Ⅰ的主要单体(DTM-B-1),纯度≥80%,因样品量少,适宜用Pre-HPLC 进一步纯化。按照“2.5”项条件用20%甲醇等度洗脱,收集24.00~27.50 min 时的流出液(见图3A),采用“2.2”项方法检测纯度。根据峰面积归一化计算纯度(见图3B),可得化合物Ⅰ(纯度96.9%),重复进行以上化合物Ⅰ的收集(54 mg)。

图3 化合物Ⅰ制备液相纯化(A)及纯化后(B)的色谱图Fig.3 Chromatograms of compound ⅠPre-HPLC(A)and HPLC(B)
3.3 HSCCC溶剂系统的选择
根据HSCCC 理论,选择一个合适的溶剂体系是能否有效分离目标化合物的关键。用于HSCCC分离的溶剂体系除能够很好的溶解样品外,还需具备目标化合物在两相中能适当分配(0.5<<2)、两相能快速分层(<30 s)及固定相能较好的保留(60%>)等条件。分配系数(值)过小,样品随流动性迅速流出,不能有效分离组分;而值过大则延长出峰时间,峰形变异。根据待分离组分的极性,本实验选择4 种不同极性的溶剂体系,分别测定了DTM-B-2 中2 种目标化合物在其不同体积配比中的值(见表1)。从表1可知,目标化合物在(正己烷)∶(乙酸乙酯)∶(甲醇)∶(水)=1∶12∶1∶12和(石油醚)∶(乙酸乙酯)∶(甲醇)∶(水)=1∶12∶1∶12 体系中的值在0.69~2.70,且分离度()均大于1.5。考虑到石油醚极易挥发,不易控制溶剂系统的体积配比,因此选择(正己烷)∶(乙酸乙酯)∶(甲醇)∶(水)=1∶12∶1∶12为两相溶剂体系。
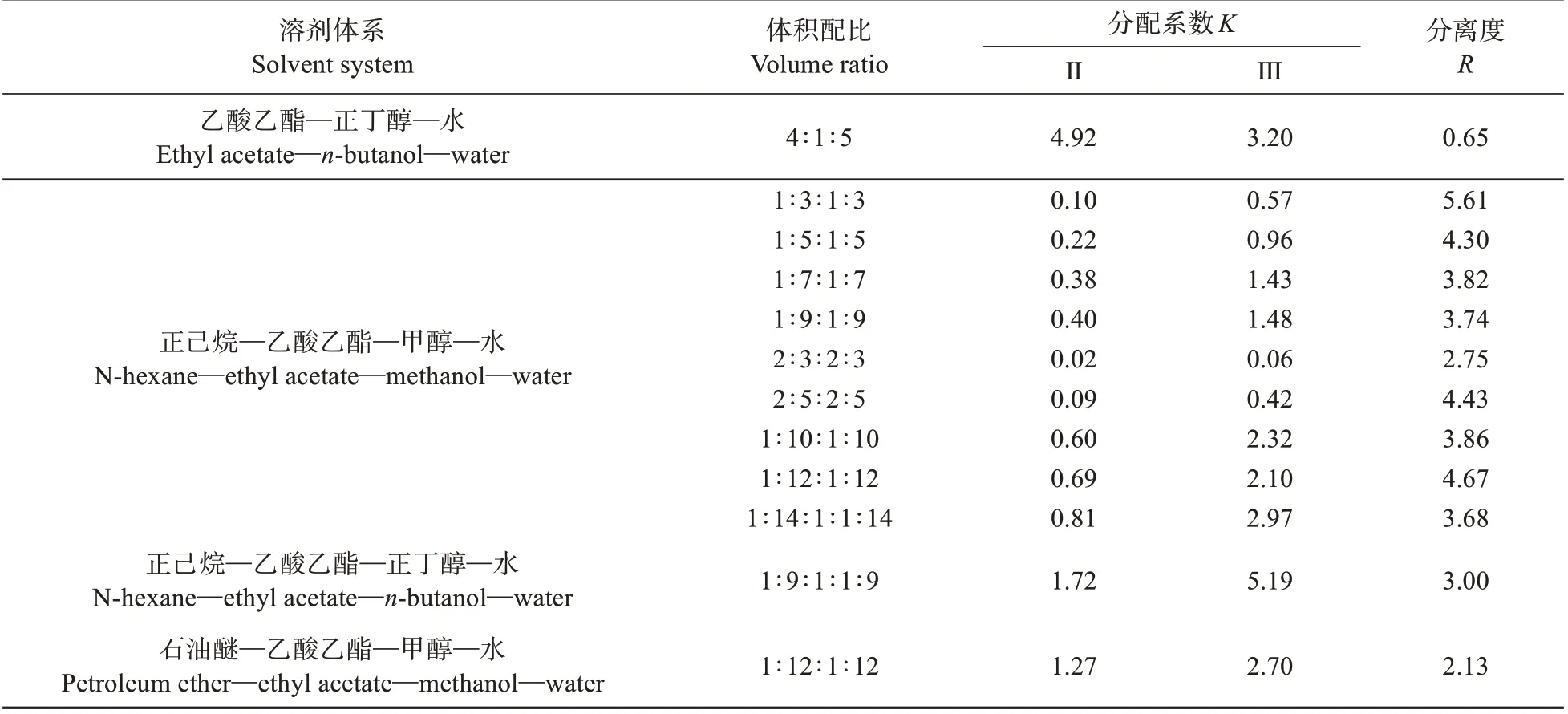
表1 目标物在不同溶剂体系中的分配系数Table 1 Partition coefficient of target compound in different two-phase solvent systems
3.4 HSCCC分离条件的优化
逆流分离时,温度对目标物在溶剂体系中的溶解和分配度,以及固定相的保留率均有较大影响。但当温度过高时,会使低沸点组分溶剂气化而产生气泡,影响出峰及仪器寿命。同时,主机转速对分离效果也具有较大影响,加快主机速度可提高固定相在管内的保留率,但转速过快会缩短设备寿命。此外,本实验考察了进样量(50、100、150、200 mg)对分离效果的影响,当上样量大于150mg时,因样品不能充分溶解于两相溶剂中而使分离效果下降。综合考虑上述因素对逆流分离效果的影响,本实验将HSCCC分离条件确定为:恒温装置温度25 ℃,主机转速850 r·min,单次进样量150 mg。
3.5 HSCCC分离
称取150 mg DTM-B-2,按照“2.4.2”项方法进行分离,收集组分1(DTM-B-2-1)和组分2(DTM-B-2-2)的流出液(见图4A)。采用“2.2”项HPLC 方法对两个组分进行纯度检测(见图4:B~C)。可得:化合物Ⅱ(DTM-B-2-2)和化合物Ⅲ(DTM-B-2-1)的主要单体,纯度分别为87.2%和88.2%。重复进行以上组分1 和组分2 的收集,分别获得0.5 g DTMB-3-1和0.3 g DTM-B-3-2,供进一步纯化。

图4 DTM-B-3高速逆流分离(A)和逆流分离得化合物Ⅱ(B)、Ⅲ(C)的色谱图Fig.4 Chromatograms of HSCCC separation DTM-B-3(A)and compound Ⅱ(B),Ⅲ(C)
3.6 Pre-HPLC进一步纯化
3.6.1 化合物Ⅱ的纯化
经“3.5”项分离可得到化合物Ⅱ的主要单体,纯度高于85%,采用Pre-HPLC 进一步纯化。按照“2.5”项条件用25%甲醇等度洗脱,收集22.00~25.00 min时的流出液(见图5A),采用“2.2”项方法检测纯度。根据峰面积归一化计算纯度(见图5B),可得化合物Ⅱ(纯度97.9%),重复进行以上化合物Ⅱ的收集(130 mg)。

图5 化合物Ⅱ制备液相纯化(A)及纯化后(B)的色谱图Fig.5 Chromatograms of compound ⅡPre-HPLC(A)and HPLC(B)
3.6.2 化合物Ⅲ的纯化
经“3.5”项分离可得到化合物Ⅲ的主要单体,纯度高于85%,采用Pre-HPLC 进一步纯化。按照“2.5”项条件用30%甲醇等度洗脱,收集24.00~27.50 min时的流出液(见图6A),采用“2.2”项方法检测纯度。根据峰面积归一化计算纯度(见图6B),可得化合物Ⅲ(纯度95.1%),重复进行以上化合物Ⅲ的收集(200 mg)。

图6 化合物Ⅲ制备液相纯化(A)及纯化后(B)的色谱图Fig.6 Chromatograms of compound ⅢPre-HPLC(A)and HPLC(B)
3.7 结构鉴定
化合物Ⅰ 棕褐色粉末,分子式为CHO;UV(MeOH):217,232,324 nm;H-NMR(600 MHz,MeOD):δ7.07(1H,d,=1.5 Hz,2′-H),6.80(1H,d,=8.2 Hz,5′-H),6.97(1H,dd,=8.2,1.5 Hz,6′-H),7.58(1H,d,=15.9 Hz,7′-H),6.29(1H,d,=15.9 Hz,8′-H),2.18~2.25(2H,m,2-H),5.36(1H,td,=9.0,4.3 Hz,3-H),3.75(1H,dd,=8.5,2.8 Hz,4-H),4.19(1H,td,=6.0,3.2Hz,3-H),2.13~1.96(m,2H,6-H);C-NMR(151 MHz,MeOD)δ74.96(C-1),37.54(C-2),72.20(C-3),70.62(C-4),70.05(C-5),36.86(C-6),175.97(7-C),126.41(C-1′),115.09(C-2′),145.40(C-3′),148.17(C-4′),113.84(C-5′),121.59(C-6′),145.69(C-7′),115.09(C-8′),167.33(C-9′)。以上H-NMR和C-NMR数据与文献报道的绿原酸一致。
化合物Ⅱ 淡黄色粉末,分子式为;UV(MeOH)λ:272,347 nm;H-NMR(600 MHz,DMSO):δ13.03(1H,s,5-OH),9.95(1H,s,4′-OH),9.34(1H,s,3′-OH),7.41(2H,m,2′,6′-H),6.86(1H,d,J=8.2 Hz,5′-H),6.83(1H,s,8-H),6.70(1H,s,3-H),4.93(1H,d,J=7.1 Hz,1″-H),4.08~4.02(6H,m,糖基质子),3.85(3H,s,7-OCH),1.81(3H,s,2‴-CH);C-NMR(151MHz,DMSO)δ164.71(C-2),103.18(C-3),182.59(C-4),152.30(C-5),128.37(C-6),159.18(C-7),91.84(C-8),153.22(C-9),105.27(C-10),56.94(7-OCH),121.93(C-1′),114.01(C-2′),146.26(C-3′),150.30(C-4′),116.43(C-5′),119.54(C-6′),102.54(C-1″),74.27(C-2″),76.78(C-3″),70.52(C-4″),74.48(C-5″),63.69(C-6″),170.45(C-1‴),20.92(C-2‴)。以上H-NMR 和C-NMR 数据与文献报道的胡麻甙-6″-乙酯一致。
化合物Ⅲ 白色无定形粉末,分子式为CHO;UV(MeOH):270,354 nm;H-NMR(600 MHz,MeOD):7.01(1H,d,=1.8 Hz,2-H),6.74(1H,d,=8.2 Hz,5-H),6.91(1H,dd,=8.2,1.6 Hz,6-H),7.51(1H,d,=15.9 Hz,7-H),6.23(1H,d,=15.9 Hz,8-H),6.72(1H,d,=1.6 Hz,2′-H),6.66(1H,d,=8.0 Hz,5′-H),6.58(1H,dd,=8.1,1.7 Hz,6′-H),5.15(1H,dd,=8.4,4.2 Hz,8′-H),3.06(1H,dd,=14.3,4.2 Hz,7′-H),2.97(1H,dd,=14.3,8.5 Hz,7′-H);C-NMR(151 MHz,MeOD)129.89(C-1),115.80(C-2),146.72(C-3),150.30(C-4),118.16(C-5),123.75(C-6),148.29(C-7),115.02(C-8),169.07(C-9),128.25(C-1′),122.39(C-2′),145.84(C-3′),147.38(C-4′),117.09(C-5′),116.88(C-6′),38.51(C-7′),75.28(C-8′),174.22(C-9′)。以上H-NMR 和C-NMR 数据与文献报道的迷迭香酸一致。
4 结论
本文通过高速逆流色谱和制备高效液相色谱优化了从甘青青兰中同时分离制备绿原酸、胡麻甙-6″-乙酯和迷迭香酸的条件,并成功结合两种技术,从甘青青兰乙酸乙酯萃取物中制备得54 mg 绿原酸、130 mg 胡麻甙-6″-乙酯和200 mg 迷迭香酸,纯度分别为96.9%、97.9%和95.1%,3 种化合物的C-NMR、H-NMR数据与文献报道一致。所建立的方法可得较高纯度、高产率的绿原酸、胡麻甙-6″-乙酯和迷迭香酸,操作简便,更省时间,为绿原酸、胡麻甙-6″-乙酯和迷迭香酸对照品的制备及工业生产提供参考,也为其药理活性的深入研究提供基础。
