PDMS/GO@Fe3O4海绵顶空固相萃取结合GC-MS分析薰衣草中挥发性成分
2022-01-25王丽丽王彩娟符继红
刘 欣,王丽丽,王彩娟,符继红
(新疆大学石油天然气精细化工教育部重点实验室,新疆 乌鲁木齐 830046)
薰衣草(LavandulaangustifoliaMill)是唇型科薰衣草属植物[1],大面积种植于新疆伊犁地区。薰衣草精油是薰衣草花穗的提取物,广泛应用于日用品、化妆品和食品中,是薰衣草主要的经济产品。此外,薰衣草精油还是一种传统的中草药,具有抑菌、抗焦虑、镇静催眠等作用,收载于卫生部《药品标准》中[2-3]。
薰衣草精油挥发性成分极为复杂,在植物体内含量较低。传统的提取方法,如水蒸气蒸馏法(HD)和溶剂萃取法[4-6]可靠性较高,但存在提取时间长、样品用量大、提取效率低和有毒溶剂残留等缺点[7-8]。顶空固相微萃取(HS-SPME)因其极大地减少了样品量和复杂基质干扰,具有耗时短、富集效率高等优点,现被广泛用于植物精油挥发性成分的提取[9-11]。SPME技术的关键在于选择石英纤维(吸附剂)进行吸附、富集样品中的待测物质。但是,固相微萃取装置价格昂贵、石英纤维易破损且纤维头材料的选择有限,限制了SPME的使用。因此,制备性质稳定、价格低廉、高性能的新型吸附和富集材料,对开发高效的天然产物精油提取分离方法具有重要意义。聚二甲基硅氧烷(PDMS)是以硅氧结构为主链构成的有机硅高分子聚合物,其兼具无机和有机材料的特性,对有机物具有良好的亲和力。海绵状的PDMS材料具有更大的比表面积和更强的吸附能力。石墨烯及其衍生物作为良好的吸附材料,其表面积大、表面活性位点多、成本低廉,被认为是有潜力的吸附材料[12-14]。四氧化三铁(Fe3O4)具有粒径小、比表面积大、活性位点多等优点,被广泛应用于样品前处理技术中[15-16]。
本研究拟制备新型PDMS/GO@Fe3O4海绵吸附材料,实现多种材料优势的叠加;建立微波辅助HS-PDMS/GO@Fe3O4萃取结合GC-MS法测定不同年份采摘的薰衣草挥发性成分;同时利用主成分分析法(PCA)对薰衣草挥发性成分进行分析,并区分不同采摘批次的薰衣草样品。
1 实验部分
1.1 仪器与试剂
GCMS-QP2010 Ultra气相色谱-串联质谱仪:日本Shimadzu公司产品;SU8010扫描电子显微镜(SEM)、H-600透射电子显微镜(TEM):日本Hitachi公司产品;VERTEX 70型傅里叶变换红外光谱(FTIR)仪:德国Bruker公司产品。
氧化石墨烯(GO):南京XFNANO材料科技有限公司产品;PDMS预聚体(Sylgard 184A)、固化剂(Sylgard 184B)、二甲基硅油:美国Dow Corning公司产品;乙酰丙酮铁(Ⅲ)、乙二醇:均为分析纯,上海阿拉丁生化科技有限公司产品;氯化钠、乙醇、正己烷、丙酮、二氯甲烷、乙酸乙酯:均为分析纯,天津致远化学试剂有限公司产品;C5~C24正构烷烃标准品:色谱纯,上海试剂一厂产品。
薰衣草:从新疆伊犁种植基地采摘的法国蓝品种,采摘时间分别为2012年和2017年,在-20 ℃密封保存。
1.2 GO@Fe3O4的制备
根据文献[17]报道,合成了GO@Fe3O4纳米颗粒。称取30 mg氧化石墨烯和100 mg乙酰丙酮铁(Ⅲ),加入27.5 mL乙二醇,超声30 min后移入高压釜中,在180 ℃下反应16 h,分别用去离子水和无水乙醇冲洗反应产物,最后将所得产物在60 ℃下真空干燥3 h,得GO@Fe3O4。
1.3 PDMS/GO@Fe3O4的制备
分别称取1.5 g Sylgard 184A、0.15 g Sylgard 184B和5 mL二甲基硅油,混合后超声,形成均匀溶液。称取2.4 mg GO@Fe3O4和3.0 g NaCl,混合均匀后加入上述混合溶液中,以8 000 r/min离心20 min,将沉淀物在80 ℃固化12 h。随后将反应产物浸泡在40 ℃水中,以溶解NaCl微粒,再用二氯甲烷和乙醇洗涤3次,最后在60 ℃下真空干燥3 h,制备得PDMS/GO@Fe3O4海绵。对2.1、2.4、2.8、3.3 mg GO@Fe3O4负载量进行优化,合成GO@Fe3O4负载量不同的PDMS/GO@Fe3O4海绵。根据文献[18]方法制备没有负载GO@Fe3O4的PDMS。
1.4 实验条件
1.4.1色谱条件 Agilent DB-5MS色谱柱(30 m×0.25 mm×0.25 μm);程序升温:初始温度50 ℃,保持1 min,以3 ℃/min升至200 ℃;进样量1 μL,分流进样,分流比5∶1;载气为氦气,流速1.0 mL/min。
1.4.2质谱条件 电子轰击(EI)离子源,电离能量70 eV,传输线温度250 ℃,离子源温度230 ℃,溶剂延迟5 min,质量扫描范围m/z30~500。
1.5 微波辅助HS-PDMS/GO@Fe3O4萃取
称取10 mg薰衣草干花和10 mg PDMS/GO@Fe3O4海绵于顶空瓶中。将顶空瓶放入微波萃取仪中,在600 W微波功率下萃取10 min。萃取结束后,将PDMS/GO@Fe3O4海绵从顶空瓶中取出,放入PE管中,随后加入200 μL正己烷超声5 min。最后,使用微量注射器移取1 μL正己烷,注入GC-MS进行分析。
2 结果与讨论
2.1 材料表征
2.1.1透射电镜和扫描电镜 采用透射电镜和扫描电镜对GO@Fe3O4和PDMS/GO@Fe3O4海绵的结构和形貌进行表征,结果示于图1。GO@Fe3O4复合材料的TEM图示于图1a。可见,在GO的二维片层材料上分布着尺寸规整的Fe3O4纳米颗粒,由于GO的片层很薄,在TEM电镜中几乎为透明的,并具有明显的褶皱。Fe3O4纳米颗粒粒径约为100 nm。GO@Fe3O4复合材料的SEM图示于图1b。可见,Fe3O4纳米颗粒紧密负载于氧化石墨烯片层上,呈现不规则形状,而且颗粒尺寸均处于纳米尺寸范围内。复合材料呈现无规则的团簇结构,验证了石墨烯作为基体形成了多个成核点,促进了Fe3O4分散生长。PDMS/GO@Fe3O4的SEM图示于图1c,其呈现宏观孔隙的分层多孔三维互联结构,表面均匀分布GO@Fe3O4颗粒突起,使PDMS/GO@Fe3O4海绵的骨架表面变得更加粗糙,获得了更大的比表面,提高了复合材料的疏水性。

图1 GO@Fe3O4透射电镜图(a),GO@Fe3O4(b)和PDMS/GO@Fe3O4(c)的扫描电镜图Fig.1 Transmission electron microscopy of GO@Fe3O4 (a), scanning electron microscopy of GO@Fe3O4 (b) and PDMS/GO@Fe3O4 (c)


图2 PDMS,GO@Fe3O4和PDMS/GO@Fe3O4的红外光谱(a)和拉曼光谱(b)Fig.2 FTIR (a) and Raman (b) spectra of PDMS, GO@Fe3O4 and PDMS/GO@Fe3O4
GO@Fe3O4的拉曼光谱在1 351 cm-1(D峰)和1 597 cm-1(G峰)上显示了2个突出的峰,示于图2b。PDMS在488 cm-1和708 cm-1处出现的峰分别对应于Si—O—Si和Si—C的拉伸振动,其他峰值出现在~2 905 cm-1和~2 965 cm-1处,对应于PDMS中—CH3基团的对称和非对称振动。与PDMS光谱相比,由于基质中存在GO@Fe3O4,PDMS/GO@Fe3O4海绵光谱中的特征峰强度增强,复合材料G峰没有发生变化,表明PDMS在聚合过程中对GO@Fe3O4的微观结构没有影响。
2.2 PDMS/GO@Fe3O4的重现性
本实验考察了PDMS/GO@Fe3O4海绵制备的重现性。使用3批独立制备的PDMS/GO@Fe3O4对薰衣草中挥发性化合物进行提取。萃取后,将PDMS/GO@Fe3O4解吸,并对萃取剂进行GC-MS分析。计算PDMS/GO@Fe3O4萃取的芳樟醇、萜品烯-4-醇、乙酸芳樟酯、乙酸薰衣草酯、石竹烯和石竹烯氧化物6种代表性化合物的重现性,其相对标准偏差(RSD)分别为2.42%、3.78%、1.59%、3.67%、4.33%和3.40%,表明PDMS/GO@Fe3O4海绵具有良好的重现性。
2.3 HS-PDMS/GO@Fe3O4萃取条件的优化
为了获得最佳的萃取效率,本实验对微波辅助HS-PDMS/GO@Fe3O4的萃取条件进行优化。选取薰衣草精油中芳樟醇、萜品烯-4-醇、乙酸芳樟酯、乙酸薰衣草酯、石竹烯和石竹烯氧化物6个代表性挥发性化合物的峰面积和薰衣草挥发性化合物的总峰面积为考察指标,平行实验3次。
2.3.1不同GO@Fe3O4负载量的PDMS/GO@Fe3O4海绵 GO@Fe3O4在PDMS/GO@Fe3O4海绵材料中负载量的大小直接影响萃取效率。本实验考察了GO@Fe3O4负载量分别为2.1、2.4、2.8、3.3 mg时,制备的PDMS/GO@Fe3O4海绵对薰衣草挥发性化合物提取效率的影响,结果示于图3。可知,GO@Fe3O4负载量的变化对PDMS/GO@Fe3O4海绵萃取效率影响较大。在GO@Fe3O4负载量由2.1 mg 增大到2.4 mg时,6个薰衣草代表性化合物的峰面积和总峰面积均达到最大值。随着GO@Fe3O4负载量从2.4 mg增大到3.3 mg,PDMS/GO@Fe3O4海绵的萃取效率降低。这可能是因为过量的GO@Fe3O4负载量会降低PDMS海绵孔洞的比表面积,从而影响萃取效率。因此,选择2.4 mg为最佳的GO@Fe3O4负载量。

图3 GO@Fe3O4负载量对PDMS/GO@Fe3O4海绵萃取效率的影响Fig.3 Effect of GO@Fe3O4 dosage on the PDMS/GO@Fe3O4 extraction efficiency
2.3.2微波功率的影响 本实验考察了300、400、500、600、700 W微波功率对HS-PDMS/GO@Fe3O4萃取薰衣草挥发性化合物效率的影响,结果示于图4a。可见,在300~600 W微波功率时,薰衣草挥发性化合物的峰面积和总峰面积随着微波功率的增加而增加,当微波功率为600 W时,萃取效率达到最大值,随后萃取效率逐渐降低。样品温度随着微波功率的增加而增大,高温有助于薰衣草中挥发性化合物的释放,但固相萃取是放热过程,温度过高会降低HS-PDMS/GO@Fe3O4的萃取效率。因此,选择600 W为HS-PDMS/GO@Fe3O4萃取的最佳微波功率。
2.3.3微波辐射时间的影响 本实验考察了2、4、6、8、10、12 min微波辐射时间对HS-PDMS/GO@Fe3O4萃取效果的影响,结果示于图4b。可见,随着微波辐射时间从4 min增加到10 min,化合物的峰面积和总峰面积逐渐增大,当微波辐射时间为10 min时,萃取效率达到最大值;随着萃取时间从10 min延长到12 min,化合物的峰面积减小,HS-PDMS/GO@Fe3O4萃取效率降低。这是由于萃取是一个平衡过程,需要一定的时间达到萃取平衡。同时,随着萃取时间的延长,萃取体系的温度升高,有助于对挥发性化合物进行快速萃取。但是,过长的微波辐射时间会造成萃取体系的温度过高,降低了HS-PDMS/GO@Fe3O4的萃取效率。因此,选择微波辐射时间10 min。
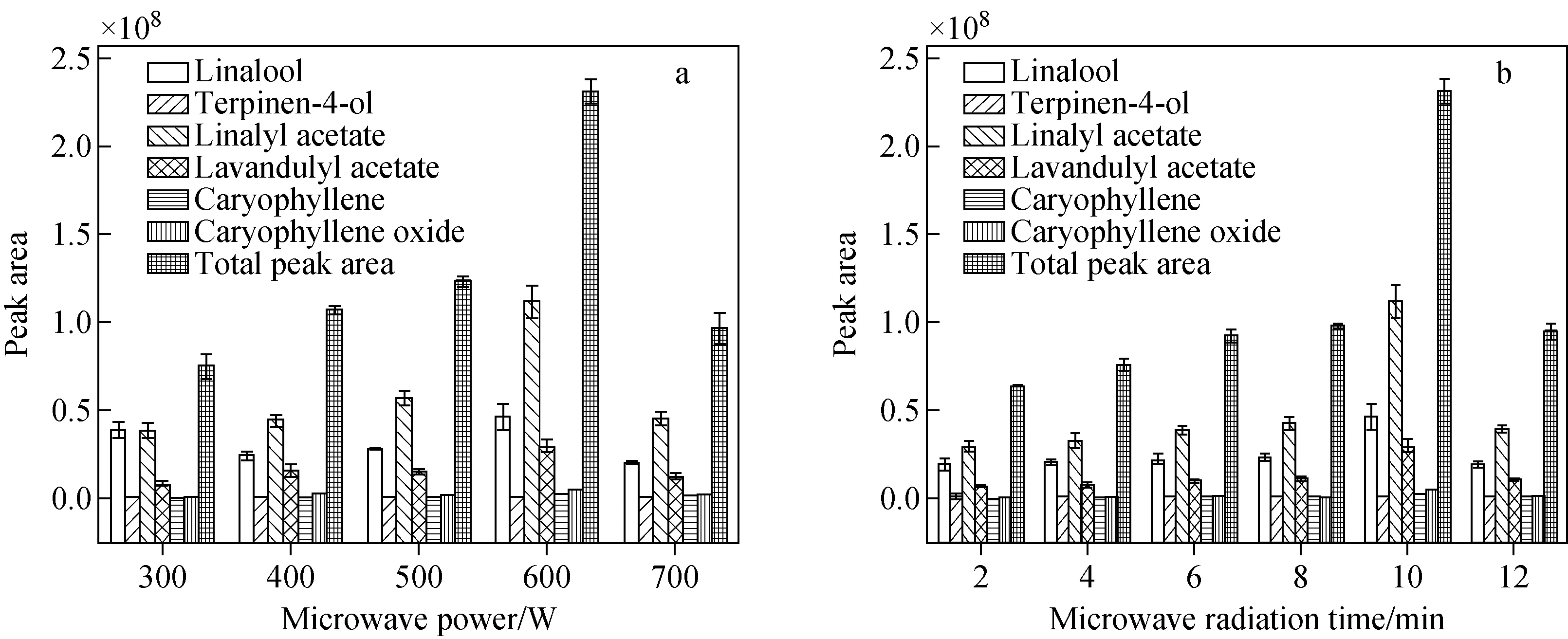
图4 微波功率(a)和微波照射时间(b)对萃取效率的影响Fig.4 Effects of microwave power (a) and microwave radiation time (b) on the extraction efficiency
2.3.4萃取溶剂的影响 萃取溶剂是萃取过程中重要的因素之一,其需要具有较好的萃取效率、环保且毒性低等特性。本实验考察了乙醇、正己烷、丙酮、乙酸乙酯等4种溶剂对PDMS/GO@Fe3O4吸附挥发性化合物萃取效率的影响,结果示于图5。可知,不同萃取溶剂对薰衣草挥发性化合物的萃取效率具有显著差异,其中萃取效率最高的是正己烷,6个代表性化合物的峰面积和总峰面积均达到最大值。因此,选择正己烷作为萃取溶剂。
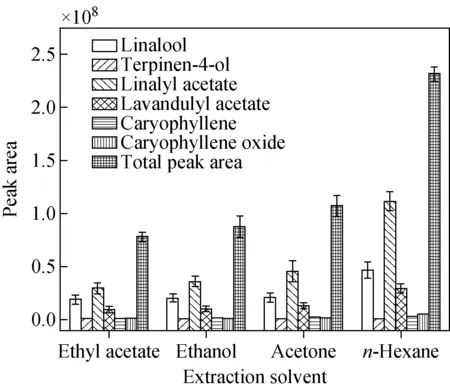
图5 萃取溶剂对萃取效率的影响Fig.5 Effect of the extraction solventson the extraction efficiency
2.4 方法验证
在HS-PDMS/GO@Fe3O4最佳萃取条件下,对线性范围、相关系数、检出限及精密度等条件进行了考察,结果列于表1。芳樟醇、萜品烯-4-醇、乙酸芳樟酯、乙酸薰衣草酯、石竹烯和石竹烯氧化物的标准曲线线性关系良好,线性范围为7.5~120 ng,相关系数(R2)大于 0.998 8,检出限(LOD)和定量限(LOQ)范围分别为0.14~0.33 ng和0.50~0.80 ng;连续进样6次,化合物峰面积的RSD均小于7%,表明HS-PDMS/GO@Fe3O4方法的重现性和精密度较好。
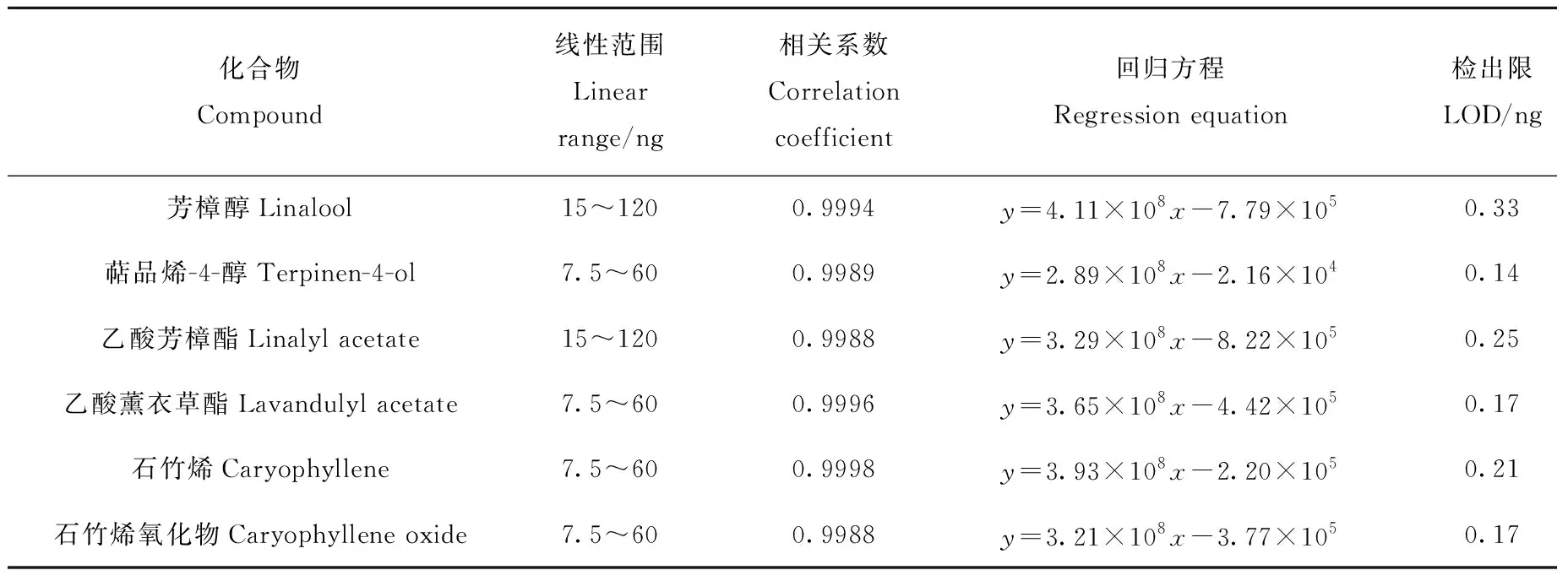
表1 方法验证Table 1 Method validation
薰衣草挥发性成分复杂,需通过样品前处理技术对其进行分离富集,再用相应的仪器进行检测分析,样品前处理已成为分析中的关键技术。近年来,为了克服传统的水蒸气蒸馏法(SD)和溶剂萃取法的样品及有机溶剂用量大、提取效率低和提取时间长等缺点,HS-SPME技术被广泛应用于天然产物挥发性成分的提取。但是,商品化的SPME技术具有纤维涂层材料种类选择有限、成本较高等缺点,限制了SPME的使用。本实验建立的HS-PDMS/GO@Fe3O4法每次分析仅需10 mg PDMS/GO@Fe3O4海绵复合材料和10 mg薰衣草样品,方法重现性好,RSD低于7%。
2.5 薰衣草挥发性成分分析
对不同采收期的18个薰衣草样品(2012年(A1~A8)和2017年(B1~B10))的挥发性化合物进行提取,其总离子流色谱图示于图6。通过GC-MS计算机谱库检索,并结合保留指数,共鉴定出52个化合物。准确称取6份B3平行样品,在最佳的萃取条件下提取分析,计算化合物峰面积的RSD值,结果列于表2。可知,薰衣草挥发性化合物以烯烃类、酯类、醇类、酮类为主,不同年份采摘的薰衣草挥发性成分及含量差异较大,但是乙酸芳樟酯和芳樟醇仍是2个主要成分,这与文献[19]报道一致。其中,2012年采摘的薰衣草主要挥发性化合物是乙酸芳樟酯(27.31%~39.87%)、芳樟醇(15.09%~20.89%)、乙酸薰衣草酯(10.26%~12.69%)、2,6-二甲基-3,7-辛二烯-2,6-二醇(5.09%~9.44%)、反式-芳樟醇氧化物(3.64%~6.79%)、环氧-α-萜烯乙酸酯(2.51%~5.44%)和石竹烯氧化物(1.06%~2.19%)等;2017年采摘的薰衣草主要挥发性化合物是乙酸芳樟酯(47.71%~56.12%)、芳樟醇(22.17%~26.67%)、乙酸薰衣草酯(5.27%~11.79%)、石竹烯(2.04%~3.61%)、顺式-芳樟醇氧化物(1.36%~1.78%)、萜品烯-4-醇(0.95%~2.16%)和(Z)-β-法呢烯(0.55%~1.20%)等。2012年采摘的薰衣草样品中,乙酸芳樟酯、芳樟醇、石竹烯的相对含量低于2017年的薰衣草样品;而2012年的薰衣草样品中,反式-芳樟醇氧化物、石竹烯氧化物和环氧-α-萜烯乙酸酯的相对含量高于2017年的薰衣草样品。其原因是乙酸芳樟酯、芳樟醇和石竹烯等单萜类化合物性质不稳定,容易发生自氧化;乙酸芳樟酯氧化生成具有多个化学官能团(羟基、氧代和环氧衍生物)的氧化产物;芳樟醇、石竹烯氧化会生成芳樟醇氧化物和石竹烯氧化物。这说明,即便采收后的薰衣草在低温环境下密封保存,但过长的保存时间仍然会导致其中不稳定化合物氧化分解。
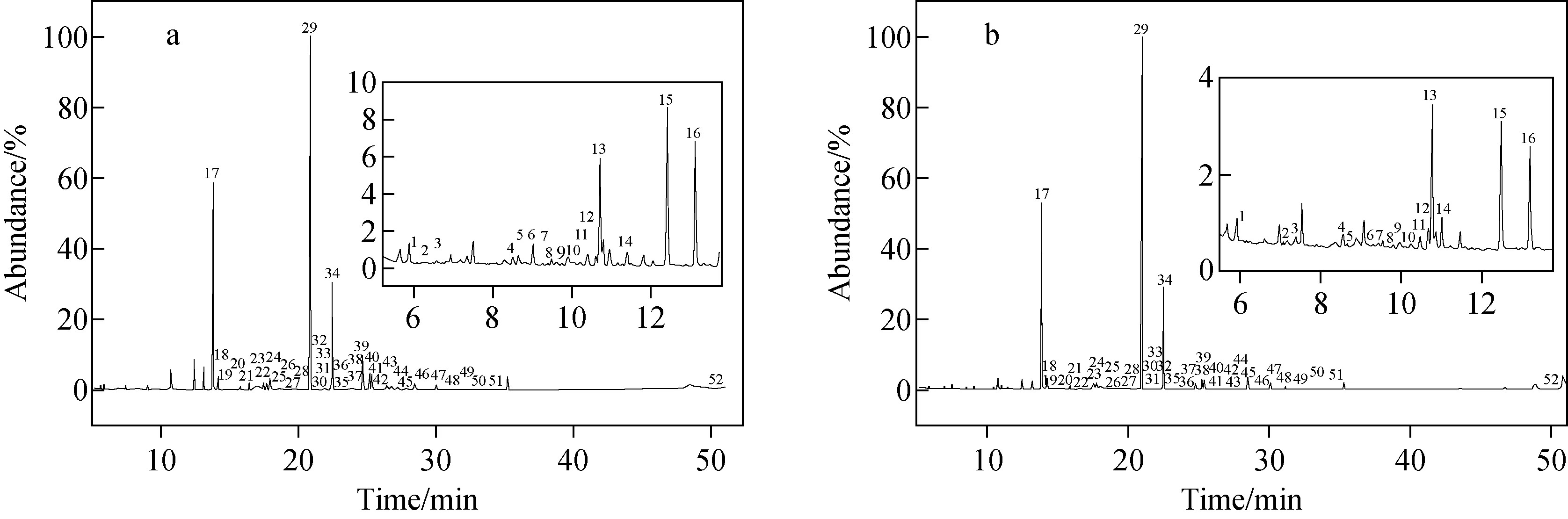
注:a.2012年;b.2017年

表2 不同采收期的薰衣草样品挥发性成分Table 2 Volatile components of lavender in different harvest years

表2

续表2
2.6 主成分分析
本工作采用主成分分析法分析不同采收批次薰衣草挥发性成分的差异,以鉴定出的52种挥发性成分峰面积为变量,对18个薰衣草样品进行分析。由分析结果可知,第一主成分(PC1)和第二主成分(PC2)的贡献率分别为70%、8.4%,表明所建立的主成分分析模型区分度较好。18个薰衣草样品根据采摘时间的不同被准确区分为2组,即2012年(A1~A8)和2017年(B1~B10),示于图7a。各变量与主成分之间的相关系数在载荷值上得以体现,距离原点较远的点权重值越大,其决定样品区分的作用就越大。对2012年分组中,贡献最大的化合物是乙酸薰衣草酯,其次为石竹烯氧化物、2-乙酰氧基-1,8-桉叶素、反式-芳樟醇氧化物和2,6-二甲基-3,7-辛二烯-2,6-二醇;对2017年分组中,贡献最大的化合物是乙酸芳樟酯,呈高度正相关,其次为(Z)-β-法呢烯、石竹烯、芳樟醇,示于图7b。

图7 主成分分析得分图(a)和载荷图(b)Fig.7 Score plot Principal component analysis (a) and loading plot (b)
3 结论
本工作制备了具有高效吸附性能的PDMS/GO@Fe3O4复合海绵材料,建立了微波辅助HS-PDMS/GO@Fe3O4结合GC-MS分析薰衣草中挥发性成分的新型样品前处理技术。该方法具有较高的灵敏度,每次分析仅需10 mg PDMS/GO@Fe3O4海绵复合材料和10 mg薰衣草样品,方法重现性好,RSD低于7%。在优化的萃取条件下,对不同年份采摘的18个薰衣草样品进行萃取分析,共确定了52个挥发性化合物。通过PCA分析,薰衣草样品可按照采摘年份的不同被准确区分。
