甘草计算机模拟指纹图谱的建立
2020-09-05顾健沛王巍李士博王小乔
顾健沛 王巍 李士博 王小乔
1. 兰州现代职业学院,甘肃 兰州730207;2. 甘肃省药品检验研究院,甘肃 兰州730030;3. 兰州市食品药品检验所,甘肃 兰州730040
甘草Glycyrrhiza uralensis Fisch.具有补脾益气、清热解毒、祛痰止咳、缓急止痛、调和诸药等功效[1-2],主要化学成分为三萜类皂苷、黄酮及多糖类成分[3-5],广泛用于医药、保健、化妆品等产品中,近20 年来,由于对野生甘草过度拆挖,甘草野生资源濒临枯竭,为满足市场的需要,国内各原甘草产区开始大量种植甘草,但对甘草的质量评价仅限于《中国药典》的质量要求,不能全信息反映甘草质量,本研究采用HPLC 检测手段,借鉴指纹图谱的研究方法,对甘肃野生与栽培的甘草通过化学成分的指纹特征进行研究,经聚类分析,形成直观、量化的数字图谱,为甘草药材的鉴定及质量控制提供了新的手段。
1 材料与方法
1.1 仪器与试药 Waters1200 液相色谱仪;岛津LC-2010A 高效液相色谱仪;SPD-M10Avp 二极管阵列测器。超声波清洗器(天津AUTOSCIENCE 公司);sartorius225D 电子天平。甘草酸单铵盐对照品(批号110731-201720),甘草苷对照品(批号11610-201607)购自中国药品生物制品检定研究院;乙腈为色谱纯,磷酸为分析纯,水为二级反渗透实验用水,色谱柱:Hypersil C18(5um,4.6mm×250mm);保护柱:Hypersil C18(5um,4mm×40mm)。甘草样品:产自甘肃省各主产地区及宁夏等地,将样品切头取除1cm 以下至0.3cm 部分。
2 方法与结果
2.1 样品来源 甘草产自甘肃省各主产地区及宁夏等地,北纬36 度线以上,13 个地区,19 批次甘草。见表1。
2.2 样品的制备 取500g 样品,粉碎成细粉,过80目筛,取约1g 甘草粉,精密称定,置100mL 容量瓶中,加80%的甲醇适量,超声提取40min,加80%的甲醇至刻度,摇匀,过滤,取续滤液用0.45μm 的微孔滤膜滤过,作为供试品溶液。取甘草酸铵对照品适量,精密称定,加甲醇制成含甘草酸铵0.2mg/mL 的溶液,即得(甘草酸重量=甘草酸铵重量/1.0207)。或按《中国药典》2015 年版方法配置。取甘草苷对照品适量,精密称定,加甲醇制成含甘草苷20μg/mL 的溶液,即得[1]。
2.3 实验方法 采用梯度洗脱的方法以A 0.5%磷酸溶液、B 乙腈,二元梯度洗脱。柱温:30℃,流速:0.8mL/min,进样量:10μL,梯度洗脱系统见表2,色谱图见图1。
《中国药典》2015 年版规定,含甘草苷(C21H22O9)不得少于0.50%,甘草酸(C42H62O16)不得少于2.0%。
2.4 甘草计算机模拟指纹图谱的建立 取上述样品,按照上述方法,对甘草样品进行处理测定,以计量标准物甘草酸保留时间为1,计算各出峰与相对保留时间的关系做各样品图,其中03 酒泉样品为不合格品,见表3,图2-6。
在前置色谱条件中,10min 前为冲洗极性糖类成分,不计入分析范围,即相对保留时间10/39=0.25,小于0.25 的计算峰不计入甘草指纹图谱相对保留时间统计。
2.5 结果 将在特征相对保留时间0.47、0.49、0.66、0.76、0.90~0.92、0.96 及1.00 峰面积归类统计,相对保留时间面积表。见表3。

表1 甘草样品采集表

表2 甘草梯度洗脱系统
3 讨论
以合格甘草为标准基样,进行分析,得出以相对保留时间为横坐标,峰面积为纵坐标的数字图谱;相对保留时间0.49 为甘草苷,相对保留时间1 为甘草酸,甘草应具备相对保留时间为0.47、0.49、0.66、0.76、0.90~0.92、0.96 及1.00 处7 个数字特征峰组成的甘草计算机模拟指纹图谱,可用于甘草全信息质量控制。
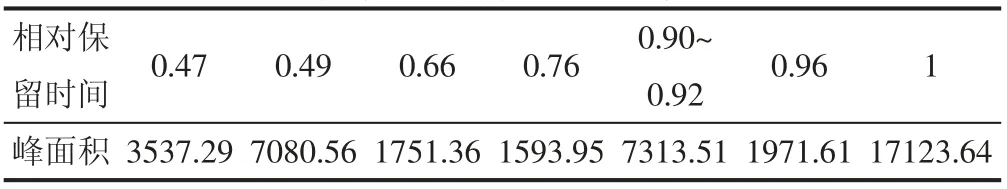
表3 甘草相对保留时间及峰面积表

图1 甘草测定指纹图谱
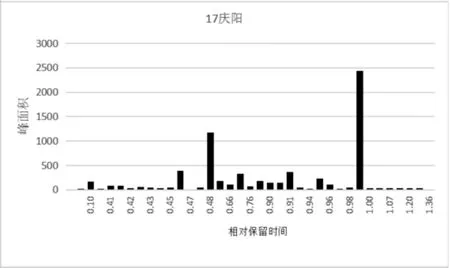
图2 样品17 庆阳(正宁)野生甘草计算机模拟指纹图谱

图3 样品4 酒泉种植4 年甘草计算机模拟指纹图谱

图4 样品19 宁夏同心种植4 年甘草计算机模拟指纹图谱

图5 样品13 通渭野生甘草计算机模拟指纹图谱
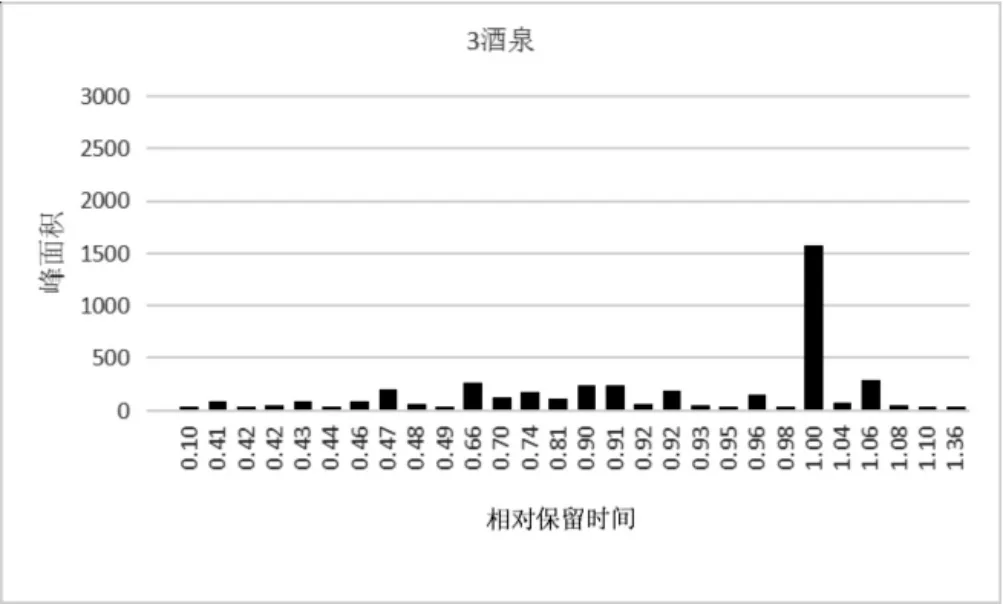
图6 样品3 酒泉种植3 年甘草计算机模拟指纹图谱
甘草应在以甘草酸为1 的保留时间,计算其他色谱峰,在相对保留时间为0.47、0.49、0.66、0.76、0.90~0.92、0.96、1.00 处有7 个特征峰。人工种植4 年甘草且种植纬度在北纬36 度以上,其甘草酸与甘草苷能同时达到《中国药典》质量要求,野生甘草与种植合格甘草有相同的数字特征峰。
