维生素E 分子印迹聚合物的二步溶胀法制备
2020-06-27朱胤霈卢轶男印建国张佑红
朱胤霈,卢轶男,印建国,张佑红*,田 莉
1.武汉工程大学化工与制药学院,湖北 武汉 430205;2.江苏玺鑫维生素有限公司,江苏 宿迁 223600
维生素E(vitamin E,VE)能促进性激素的分泌,预防流产,提高人体生育能力,市场价值潜力巨大[1]。目前我国提取的天然VE 主要来自脱臭馏出物[2],由于脱臭馏出物中含有游离脂肪酸、甾醇等杂质,提取VE 往往需要萃取、酯化、转酯化、皂化等工艺的综合运用,但这些工艺都存在溶剂成本高、资源能耗大、VE 纯度及收率低等问题[3]。因此期望寻找一种高效的分离提纯VE 的工艺过程,能够简化生产流程,降低成本。分子印迹聚合物(molecularly imprinted polymer,MIP)作用精准,能从复杂混合物中直接吸附待分离的目标物[3-6],具有优异的选择吸附性。在药物手性拆分、模拟酶催化、生物传感器等方面有很好的发展和应用[7-10]。本文选取二步溶胀聚合法[11-13],用聚苯乙烯微球作为VE 分子印迹聚合物的种球,成功合成印迹聚合物,并从吸附机理、吸附行为等方面对该聚合物进行分析研究。此研究为应用印迹聚合物提纯VE的工艺提供了一定的理论依据。
1 实验部分
1.1 化学试剂与材料
1.1.1 主要化学试剂 VE(纯度:94.02%),苯乙烯,甲基丙烯酸,正辛醇,乙二醇二甲基丙烯酸酯,聚乙烯吡咯烷酮,偶氮二异丁腈,十二烷基硫酸钠,聚乙烯醇,邻苯二甲酸二丁酯,甲苯,三氯甲烷等试剂均为分析纯。
1.1.2 主要实验仪器 旋转蒸发仪(RE-52AA,上海亚荣生化仪器厂);高效液相色谱仪(Agilent1260,安捷伦科技有限公司);索式提取器(SXT-06,上海洪纪仪器设备有限公司);恒温振荡培养箱(HZQ-X100,金坛市杰瑞尔电器);超声波细胞粉碎仪(JY92-2D,宁波新芝生物科技有限公司)。
1.2 实验方法
1.2.1 试剂前处理 苯乙烯:在分液漏斗中倒入50 mL 苯乙烯,用质量分数10%的氢氧化钠萃取3次(去除阻聚剂对苯二酚),再用超纯水反复洗涤至流出液呈中性后,将其减压蒸馏得到精制产物,低温保存。
二甲基丙烯酸乙二醇酯:处理方法与苯乙烯单体相同。
偶氮二异丁腈:取4 g 偶氮二异丁腈,加入到30 mL 乙醇中并于65 ℃加热溶解,过滤后将滤液放于冰箱内结晶24 h,所得固体放入棕色瓶内低温保存。
1.2.2 聚合物微球的合成 准确量取50 mL 乙醇和50 mL 苯乙烯于150 mL 的锥形瓶中,分别加入0.2 g 偶氮二异丁腈,1 g 聚乙烯吡咯烷酮,冰浴超声10 min,然后将溶液倒入圆底烧瓶中,升温70 ℃,氮气保护反应24 h。反应结束后,过滤除去液体干燥备用。
1.2.3 聚合物的合成 1)印迹聚合物的合成:称取1 g 聚苯乙烯微球,0.2 g 十二烷基硫酸钠,30 mL超纯水,于400 W 超声15 min(室温,超声5 s/间隔5 s,以下实验步骤超声条件相同),称取0.5 mL 邻苯二甲酸二丁酯,0.1 g 偶氮二异丁腈,1 mL 甲苯,30 mL 超纯水,于150 W 超声5 min,将两混合溶液倒入一个圆底烧瓶内,室温下150 r/min 搅拌24 h。取0.43 g VE,0.1 g 十二烷基硫酸钠,3.96 g 二甲基丙烯酸乙二醇酯(ethyleneglycol dimethacrylate,EGDMA),0.52 g 甲 基 丙 烯 酸(methacrylic acid,MAA),0.4 g 聚乙烯醇,5 mL 正辛醇,10 mL 氯仿,40 mL 超纯水,于400 W 超声15 min,加入上述圆底烧瓶中继续搅拌24 h。称取0.2 g 聚乙烯醇,20 mL 超纯水,于400 W 超声10 min,加入上述圆底烧瓶中,升温70 ℃,氮气保护反应24 h。将反应结束后得到的固体颗粒置于索氏提取器中,用甲醇-乙酸溶液(V甲醇∶V乙酸=9∶1)洗涤24 h,再用纯甲醇溶液冲洗5 h,干燥后即得VE MIPs。
2)非印迹聚合物的合成:除不加入VE 外,其他步骤与上述内容相同。
1.3 分子印迹聚合分析与表征
用扫描电子显微镜观察MIPs 的表观形貌是否为球形,表面是否有孔穴;用傅里叶红外光谱仪分别测定功能单体MAA、交联剂EGDMA 及VE MIPs的红外吸收谱带,分析功能单体是否与交联剂聚合,印迹聚合物是否具有能与VE 特异性结合的官能团。
1.4 VE 的HPLC 定量方 法
采用液相色谱法来检测VE 的含量,检测条件如下:流动相为甲醇-水溶液(V甲醇∶V水=99∶1);流速为1 mL/min;进样量为20 μL;色谱柱为C18 色谱柱(4.6 mm×100 mm,5 μm);柱温为20 ℃;检测波长为UV-294 nm。
用甲醇配制浓度梯度为1 mmol/L 的VE 溶液(浓度范围为1~5 mmol/L),采用外标法,绘制VE浓度-峰面积的标准曲线。
1.5 VE 分子印迹聚合物静态吸附实验
配制初始浓度在0.3 mmol/L 至1.8 mmol/L 之间的VE 氯仿溶液各10 mol,将其分别加入含有50 mg 分子印迹聚合物的锥形瓶中。密封置于25 ℃恒温振荡箱,振荡吸附120 min 以上。吸附结束后,混合物用0.45 μm 的有机相微膜过滤,再取滤液1 mL 于干燥箱内抽干除去氯仿,所得干燥样品再用1 mL HPLC 甲醇溶解,用高效液相色谱仪进行检测并计算印迹聚合物的吸附量:

式中,Qe—印迹聚合物对VE 的吸附达到平衡时的吸附量(μmol/g);Ce—(mmol/L);V—印迹聚合物对VE 的吸附达到平衡时的体积(mL);C0—未进行吸附时VE 的浓度(mmol/L);m—吸附剂印迹聚合物的质量(g)。
作为对照,在测定分子印迹聚合物吸附等温线的同时也对非印迹聚合物的吸附等温线进行了测定,其方法与上述相同。
1.6 分子印迹聚合物吸附机理分析
为从吸附机理上更好地理解分子印迹聚合物吸附VE 的过程,分子印迹聚合物的吸附行为选用Langmuir 吸 附 模 型[14]与Freundlich 吸 附 模 型[15]来进行分析,印迹聚合物与VE 结合特性的分析选用Scatchard 模型来进行。
Langmuir 方程式为:
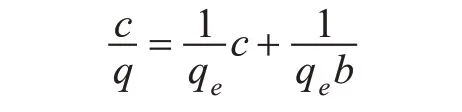
式中:c—印迹聚合物对VE 的吸附达到平衡时的浓度(mg/L);q—印迹聚合物对VE 的吸附达到平衡时的吸附量(mg/g);qe—印迹聚合物对VE的饱和吸附量(mg/g);b—吸附平衡常数。
Freundlich 方程:
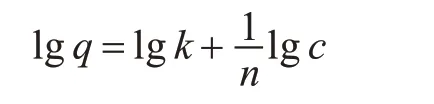
式中:q—印迹聚合物对VE 的吸附达到平衡时的吸附量(mg/g);c—印迹聚合物对VE 的吸附达到平衡时的浓度(mg/L);k 与n—特征常数。浓度对吸附能力的影响用1/n 来表示,当0.1<1/n<0.5时,吸附质容易被吸附;当1/n>2 时,吸附质不易被吸附[14]。
Scatchard 模型[16]为:
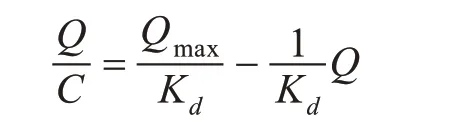
式中,C—印迹聚合物对VE 的吸附达到平衡时的浓度(mmol/L);Q—印迹聚合物对VE 的吸附达到平衡时的吸附量(μmol/g);Qmax—印迹聚合物对VE 的最大表观吸附量(μmol/g);Kd—平衡解离常数(mmol/L)。
2 结果与讨论
2.1 分子印迹聚合物的表观形貌
由图1 可知,二步溶胀法制得的分子印迹聚合物微球呈现均匀球形,外形规则、无破损,其表面存在一定的孔穴,为后续的静态吸附实验提供了良好的基础。
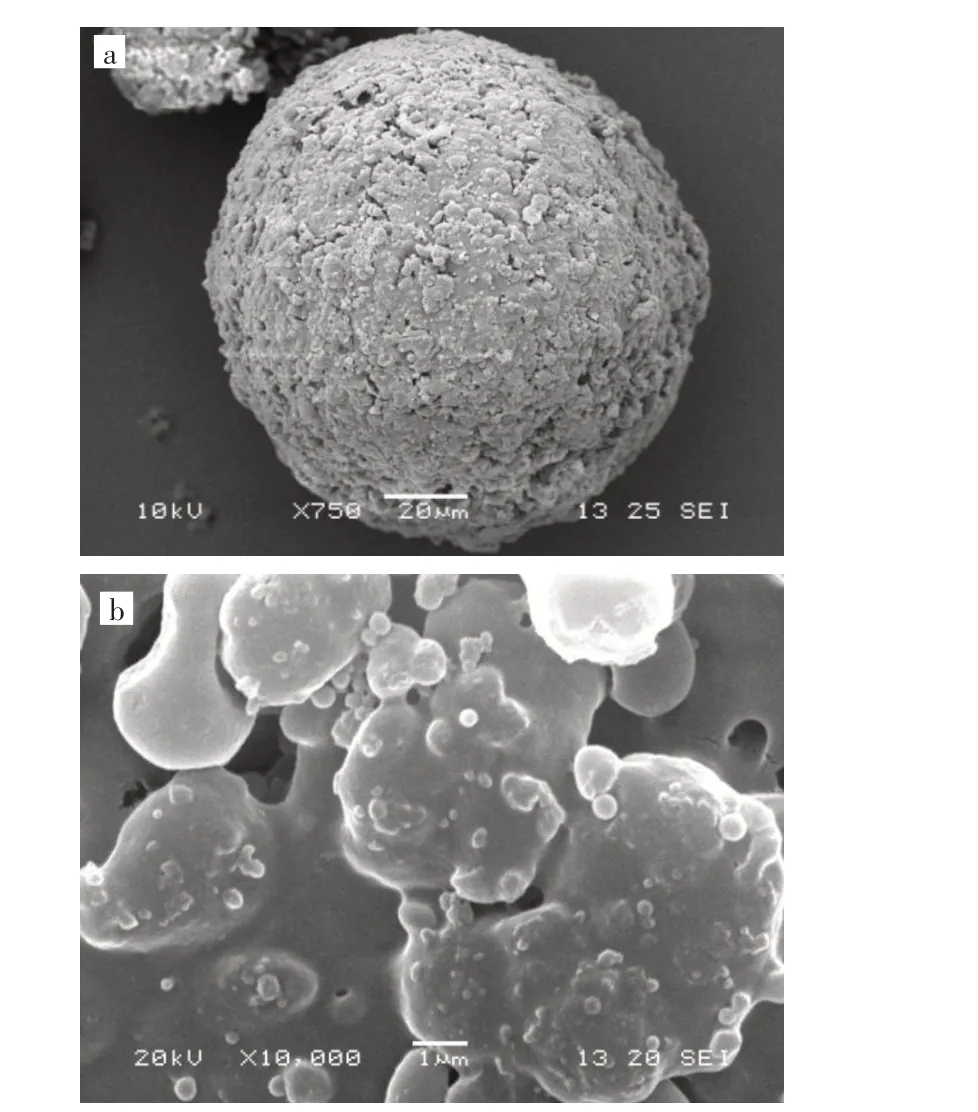
图1 分子印迹聚合物的扫描电镜图:(a)聚合物微球,(b)微球表面Fig.1 SEM images of MIPs:(a)polymer microsphere,(b)surface of the micropshere
2.2 分子印迹聚合物的红外分析
图2 中的a、b 及c 曲线分别是功能单体MAA、交联剂EGDMA 及MIPs 的红外吸收光谱图。在交联剂c 曲线红外谱图(Infrared Spectroscopy,IR)中可以看出,在3 400 cm-1处的吸收峰是羟基的伸缩振动峰,在1 720 cm-1处的吸收峰是羰基的伸缩振动峰,这两类峰的出现表明由聚合作用得到的聚合物中已经引入了可以同模板分子作用的羟基和羰基基团。1 637 cm-1附近的碳碳双键峰值较弱,且在1 150 cm-1处出现了碳氧键的伸缩振动峰,说明绝大多数的功能单体MAA 和交联剂EGDMA 已经进行交联聚合成功,仅剩部分残余未聚合。
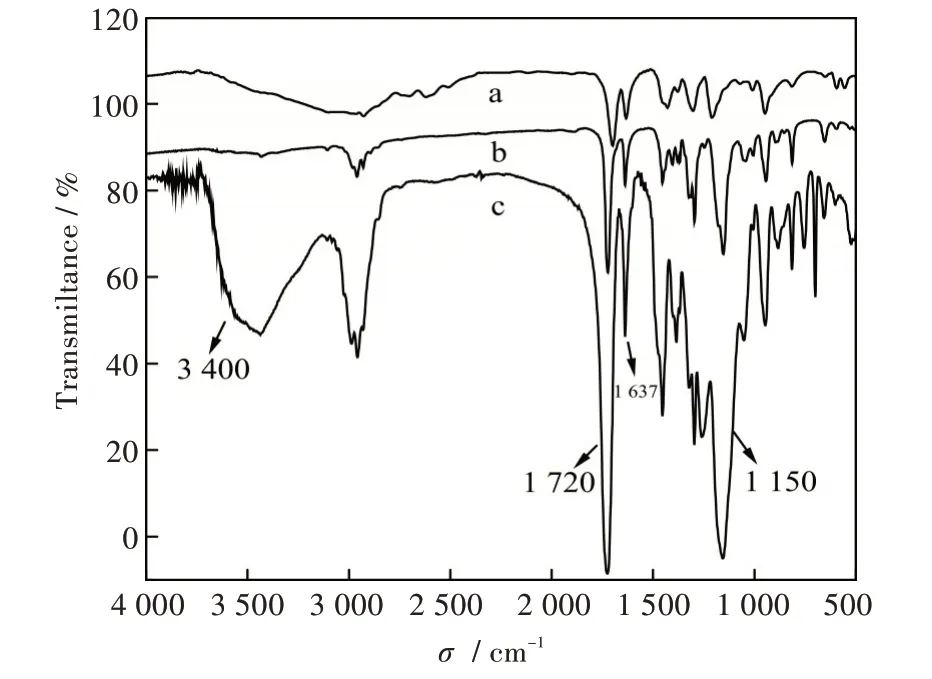
图2 (a)MAA,(b)EGDMA,(c)MIPs 的红外光谱图Fig.2 IR spectra of(a)MAA,(b)EGDMA and(c)MIPs
2.3 标准曲线的绘制
由图3 可知,在所测量的范围VE 浓度与峰面积有良好的线性关系,因此在后续的实验过程中,可以通过色谱检测得到的峰面积数据,来计算静态吸附溶液中VE 浓度的具体数值。
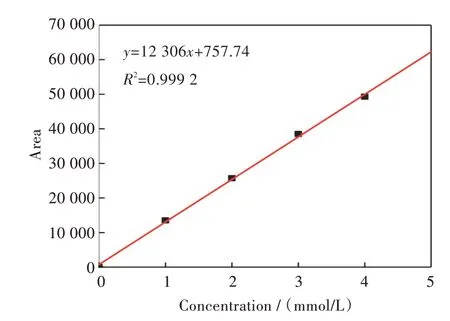
图3 VE 的标准曲线Fig.3 Standard curve of VE
2.4 吸附等温线
吸附等温线研究的是分子印迹聚合物吸附容量随VE 平衡浓度的变化情况,以吸附到达平衡时VE 的浓度为横坐标,以吸附剂的平衡吸附量为纵坐标作图可得吸附等温线,如图4 所示。当VE 初始浓度逐渐增大时,在印迹聚合物中VE 的吸附量持续增加,而非印迹聚合物对VE 的吸附量很低且很快到达平衡。这是由于功能单体与VE 通过氢键作用力形成了具有特定空间结构的孔穴及相应的结合位点,这令分子印迹聚合物与VE 之间产生了较高的亲和力,而非印迹聚合物与模板分子维生素E 的结合主要是通过无规律的物理吸附作用力,没有相对应的结合位点,吸附能力较差。
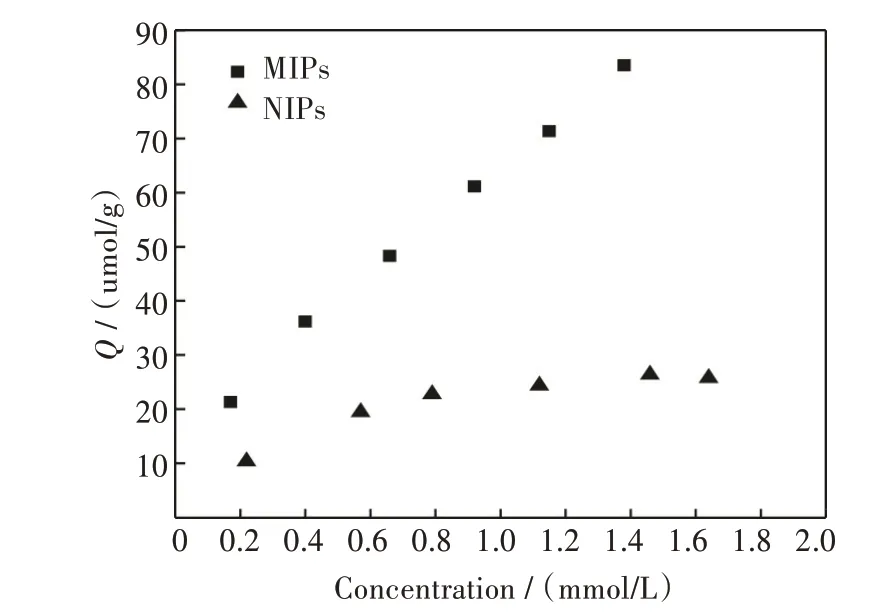
图4 分子印迹聚合物和非印迹聚合物对VE 的吸附等温线Fig.4 Adsorption isotherms of MIPs and NIPs towards VE
2.5 吸附等温线模型关联
Langmuir 线性拟合图,如图5(a)所示;Freundlich 线性拟合图,如图5(b)所示。拟合数据见表1。由拟合结果得知,Langmuir 模型拟合得出饱和吸附量是52.083 mg/g,这与通过静态吸附实验所测的最大吸附量38.12 mg/g 差别较大,说明此模型对VE 聚合物的关联效果不好。Freundlich 模型计算所得1/n 值为0.488 04,说明印迹聚合物能很好的吸附目标物,单位浓度吸附量则为1.608 mg/g。Freundlich 模型的相关系数为0.991 98,高于Langmuir 模型,这表明Freundlich 模型更适用于VE 印迹聚合物吸附量数据的关联。
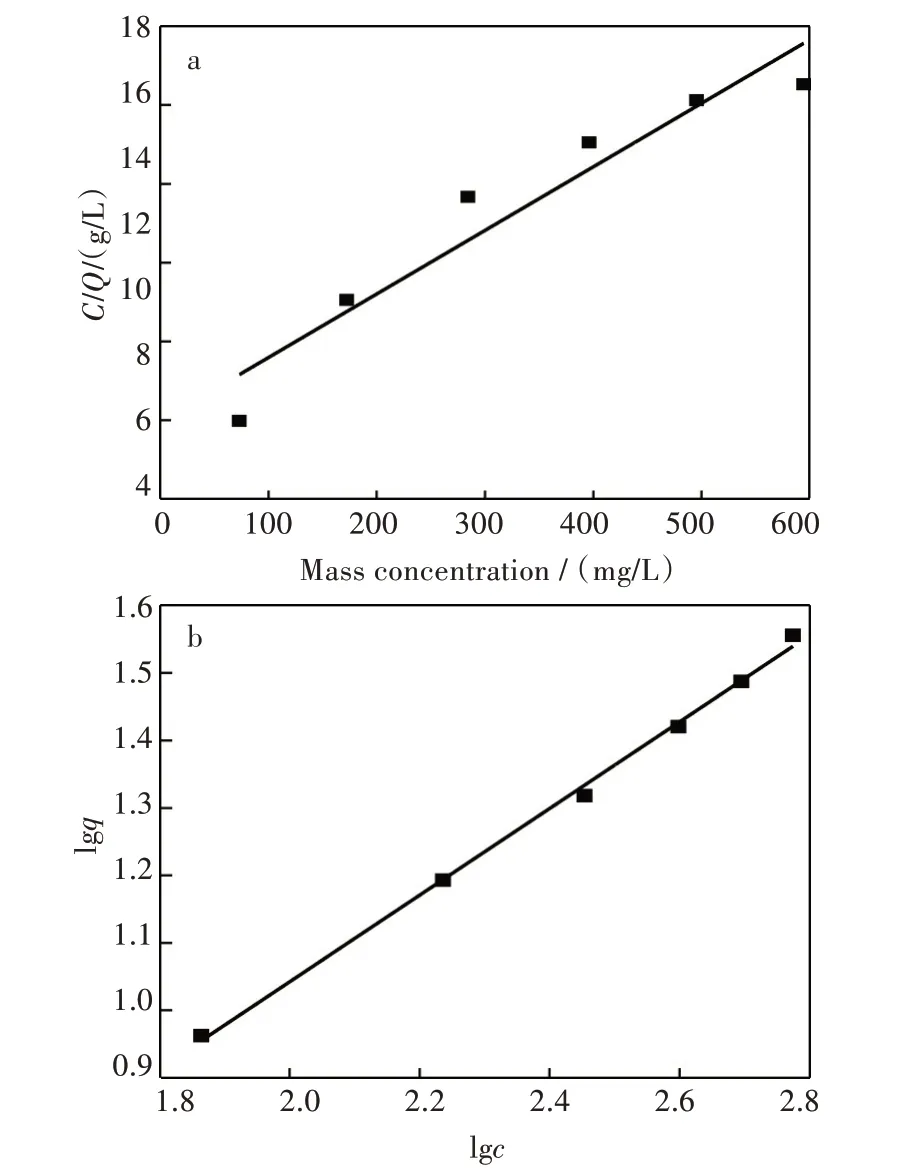
图5 吸附等温线拟合图:(a)Langmuir,(b)FreundlichFig.5 Fitting plots of adsorption isotherm:(a)Langmuir equation,(b)Freundlich equation
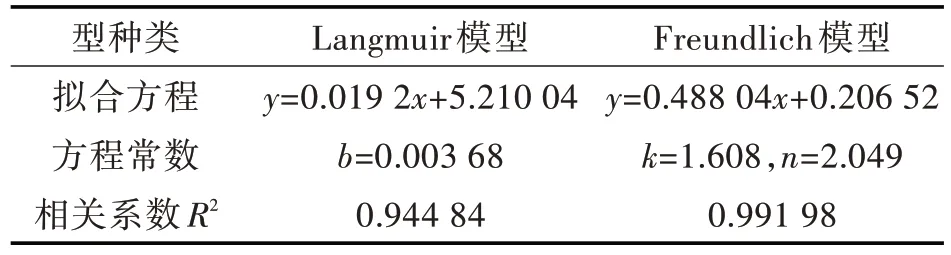
表1 印迹聚合物和非印迹聚合物的吸附等温模型拟合参数Tab.1 Isotherm model parameters for adsorption of MIPs towards VE
2.6 Scatchard 分析
印迹聚合物与非印迹聚合物的Scatchard 曲线图如图6 所示,图中印迹聚合物的Scatchard 曲线形成了两个不同的部分,对其分别进行线性拟合,计算解离常数和最大表观吸附量。具体数据可见表2。
由Scatchard 曲线拟合结果可以得知在VE 印迹聚合物中存在两种结合位点,一种特异性结合位点,另一种是非特异性的结合位点。在VE 初始浓度较低时,印迹聚合物中的特异结合位点优先与VE 互相结合,结合较为迅速、紧密,因而Scatchard 曲线较陡,解离常数较小;而在浓度较高的条件下,印迹聚合物中的特异性结合位点快速达到平衡后非特异性结合位点逐渐发挥作用,但其对VE 的亲和力不及特异性结合位点,Scatchard曲线平缓,解离常数较大。
非印迹聚合物中只有一种结合位点,因此其拟合结果为一条直线;由于这种位点对模板分子的结合是非特异性的,Scatchard 拟合曲线斜率平缓,解离常数相对印迹聚合物较高。
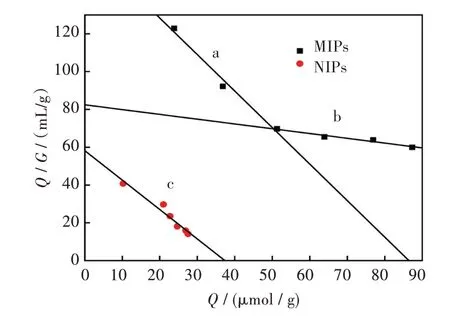
图6 印迹聚合物与非印迹聚合物的Scatchard 曲线:(a)印迹聚合物特异性结合位点,(b)印迹聚合物非特异性结合位点,(c)非印迹聚合物Fig.6 Scatchard polt of MIPs and NIPs:(a)MIPs specific binding site,(b)MIPs non-specific binding sites,(c)NIPs

表2 印迹聚合物与非印迹聚合物对维生素E 的Scatchard 拟合参数Tab.2 Scatchard fitting parameters of MIPs and NIPs towards VE
3 结 论
VE 在医药、生活等方方面面都有巨大的应用价值,传统的分离提纯方法消耗试剂多污染环境,提纯步骤复杂,本实验通过二步溶胀法合成的分子印迹聚合物,对VE 的吸附量为38.12 mg/g。静态吸附实验所得等温曲线拟合表明Freundlich 模型(R2=0.992)对分子印迹聚合物有更好的关联性;Scatchard 模型表明VE 分子印迹聚合物中有两种结合位点。应用二步溶胀法合成的VE 分子印迹聚合物具有较好的吸附性能。
随着分子印迹技术的发展,将分子印迹聚合物用于结构相似物的分离纯化,特别是与色谱分析和吸附分离过程、固相萃取等领域已成为研究的热点。本文的工作为进一步研究VE 印迹聚合物吸附分离性能提供了一定的实验基础和理论基础,期望分子印迹技术能成为工业分离纯化VE 的一种有效手段。
