乙肝性肝纤维化发病机理研究进展
2019-03-07,
,
乙型肝炎病毒性肝炎是引起肝纤维化的重要因素,我国是乙型肝炎病毒感染的重灾区。深入研究乙肝性肝炎、肝纤维化的发病机理,找到有效防治肝纤维化的方法或药物,具有重要的社会价值和经济价值。本文就国内外近年来对乙肝性肝纤维化的研究成果进行综述,为开发有效防治乙肝性肝纤维化的新药或治疗方法提供一些可供参考的思路。
1 乙肝病毒感染与慢性肝炎、肝纤维化的发病率
我国的肝硬化多由乙型肝炎病毒、丙型肝炎病毒感染引起肝炎所致。估计全球大约有30%的人口与乙肝病毒 (HBV)接触,其中有3.5亿为HBV携带者,每年有100万人口死于与HBV相关的疾病。乙肝患病率在0.1%~20%之间,西欧国家、美国、澳大利亚、新西兰以及加拿大的患病率较低(0.1%~2%),地中海国家、日本、中亚、拉丁南美洲以及中东次之((3%~5%) ,而东南亚、中国以及撒哈拉以南非洲的患病率最高(10%~20%),疫苗的使用使其发病率大大降低,但HBV相关并发症:肿瘤以及死亡人数却仍在增加。慢性HBV感染的自然过程由病毒复制与机体免疫反应所决定,以下这些因素也可能与乙肝相关疾病的进程有关:性别、酒精消耗量以及与其它肝炎病毒共存现象等。
大约有40~50% 男性以及15%的女性会死于肝病相关的疾病,免疫活化可引起肝病进展,五年疾病进展的比例分别为:10~20%由慢性肝炎发展为肝硬化,20~30%由肝硬化代偿期发展为失代偿期,5~15%由肝硬化代偿期发展为肝细胞肝癌。据此,其生存率分别为:代偿性肝硬化5年生存率为85%;失代偿期肝硬化的1年生存率为55~70%,5年生存率为15~35%。慢性HBV感染的结局决定于HBV复制停止时肝损伤的程度有关,当HBV复制控制后,其肝纤维化有可能反生逆转[1]。慢性肝炎的肝纤维化发生率约为60%,其中25%~40%的肝纤维化患者最终发展为肝硬化[2]。
2 乙型肝炎发病机理
HBV感染引起肝纤维化的详细机理还未得到系统阐明,但普遍认同的是HBV引起的肝损伤与机体的免疫应答密切相关。HBV是一种小分子折叠式DNA病毒,由于缺乏病毒聚合酶校正活性,在病毒复制过程中常发生核酸错误掺入所致的突变,导致HBV存在八种亚型,其中B亚型和C亚型分布在亚洲。在电镜下可见三种病毒颗粒:传染性病毒体和亚病毒颗粒。病毒膜含有三种病毒表面蛋白,分别为preS1 (或大)、preS2 (或中)和preS(或小),这些蛋白与乙肝表面抗原(HBsAg)有关。几乎所有的包膜病毒都含有来自宿主的蛋白,HBV颗粒也不例外。
当HBV病毒进入宿主体内后首先引起先天性免疫应答,这种应答常开始于感染细胞迅速诱导干扰素(IFN)α/β生成,致使大量干扰素诱导基因(ISGs)表达,后者再发挥各种细胞内抗病毒机制限制病毒产量和传播,使病理变化过程缩小。
另一方面,获得性免疫迅速启动,首先是抗原-抗体反应。一般认为中和anti-HBs抗体在HBV感染恢复中发挥着重要作用——将传染性封闭在感染的宿主体内便于排出、破坏其病毒颗粒。这些抗体还可通过阻断病毒与靶细胞的受体结合而阻止细胞发生重复感染。HBeAg是由于慢性感染患者体内HBV特异性免疫太弱以至于从感染的肝细胞不能消除HBV病毒,但足以破坏HBV感染的肝细胞并引起慢性炎症性肝病。
在HBV感染期,体液抗体与循环病毒颗粒的清除、防止病毒颗粒在宿主体内传播有关,而细胞免疫与消除感染细胞有关。控制进入体内病毒的方式既有杀细胞的,也有非杀细胞的。非杀细胞作用一般通过释放细胞因子或其它具有抗病毒作用的物质,没必要损害感染细胞;相反,以杀细胞方式抑制病原则是通过杀死感染细胞而损伤组织。对病毒抗原的耐受、病毒抑制抗原加工或呈递、感染免疫特殊位点、调节对细胞毒性介质的反应以及病毒发生变异等因素均有助于病毒感染持续化。
对HBV封膜抗原的抗体反应是T细胞依赖性过程,尽管外周血CD4+应答与病毒清除及急性肝炎有明显关系,但CD4+细胞并不直接参与病毒的清除及组织损伤。HBVCD8+T细胞在病毒清除和肝脏损伤的病理中发挥了主要作用。
活化的CD4+T细胞各亚型分别产生不同的效应因子:如Th1细胞主要分泌IFNγ、淋巴毒素α(Lfα)和IL2。Th2 细胞主要分泌IL4、IL5、IL9、IL13、IL10、IL25和双调蛋白(amphiregulin)。Th17 细胞的关键效应细胞因子包括IL17A、IL17F、IL21和IL22。Treg主要效应因子包括IL10、TGF-β和IL35。Tfh1通过分泌IFNγ促进IgG2a生成、Tfh2 分泌IL4有助于IgG1和IgE的生成,而Tfh10则通过分泌IL10促进IgA分泌[3]。
病毒感染促进多种前炎性细胞因子的分泌,引起CD8+T细胞增加,这些细胞因子包括:I型干扰素、IL-12家族成员、IL-2、IL-6、IL-7、IL-15、IL-21等,同时还包括IL-1家族成员IL-1、IL-18、IL-27及IL-33。尽管不同的病毒感染可引起不同的炎症因子,但都会通过DCs和巨噬细胞产生大量的IL-12,IL-12与I型干扰素一起作用于CD8+T细胞的分化,IL-12诱导T细胞表达T-bet,高水平的T-bet刺激生命周期短暂的效应T细胞成体终末分化,而低水平的T-bet与记忆先驱T细胞的发育有关,这种T细胞可以继续形成长寿命的记忆池。CD8+T细胞活化一方面产生大量的IFNγ抑制病毒复制,另一方面产生大量的细胞因子,发挥细胞毒性作用,对组织造成损伤[1,4,5,6]。HBV引起免疫反应的相关机制见图1。
3 乙肝性肝纤维化发病机理
在免疫引起肝纤维化的过程中,病毒抗原蛋白以及免疫活化过程中产生的大量细胞因子特别是TGFβ、IL-10以及IL-18发挥了重要作用。
HBV引起的肝脏毒性与其诸多抗原蛋白密切相关。近期研究表明病毒蛋白HBeAg可以诱导Th2免疫反应,而HBcAg可诱导Th1免疫反应,当体内Th2对HBeAg的反应强于Th1 对HBcAg的反应时,其结果将导致Th1的耗竭。Th1刺激巨噬细胞清除病毒颗粒,而Th2刺激B细胞产生免疫球蛋白粘附于病毒颗粒的表面并诱导调理素作用。每次肝炎伴随的肝细胞再生与HBVDNA碎片表达病毒克隆肝炎Bx抗原(HBxAg)的增加密切相关,肝内HBxAg与慢性肝病(CLD)的强度、进展相一致。另外,HBxAg还可阻断Fas和TNFα介导的免疫性杀病毒作用,这不仅与HBxAg阻断caspase活性有关,也与持续刺激肝脏保护作用通路密切相关,如:NF-κB、PI3K以及β-catenin,HBxAg也可通过刺激纤连蛋白(FN)的生成,并通过多种机制刺激TGFβ1的生成及活性。HBxAg通过改变多种MMPs的表达而表现出重塑细胞外基质 (ECM) 的作用,这样,HBxAg通过减少免疫介导的感染肝细胞凋亡而促进感染慢性化,通过促进EMC重构使肝纤维化及肝硬化持续进展[1]。
从图1可以发现,在乙肝的发病过程中,TGFβ参与了多个免疫环节的调控,对疾病的进程起着非常重要的作用。近年的研究成果证明TGFβ在慢性肝病引起肝纤维化的过程中发挥了关键性的作用,活化的TGFβ可促使肝星状细胞(HSCs)活化以及发生纤维化,TGFβ还可直接刺激α1、α2type(I) 前胶原、基质金属蛋白酶抑制剂(TIMP)-1、TIMP-2、纤溶酶原激活物抑制剂(PAI)-1基因表达,而α平滑肌肌动蛋白(α-SMA)和结缔组织因子(CTGF)的活化则是TGFβ依赖式的。在慢性肝损伤过程的不同阶段TGFβ可能起着不同的作用,在早期,TGF-β促进肝细胞损害的同时,引起HSC转型为肌成纤维细胞促进伤口愈合;在再生和肝细胞增殖阶段,TGF-β具有组织体积限制性抑制细胞生长的作用,通过产生TRegs控制炎症;在肝病持续及肝纤维化阶段,大量的结疤形成伤口愈合反应对肝脏产生不良影响。在癌变前期,其抑制细胞生长的作用可以控制上皮细胞增殖阻止癌变发生,但同时由于TGF-β对炎症的负调节可能抑制针对初生肿瘤的免疫反应;而在癌症期,TGF-β抑制细胞生长的作用已经消失,其另外的信号通路可能有助于肿瘤的进展及转移,而且,TGF-β对内皮细胞的促血管生成作用也可能有助于肿瘤的进展。TGFβ的促纤维化作用在许多动物模型中得到了证实,而针对TGF-β信号通路的治疗策略在动物试验模型方面取得巨大成功,但是,这些策略用于临床的效果则微乎其微,导致这种结果的原因可能是TGF-β的功能具有生物多样性的特点,同时也说明肝纤维化并非TGFβ单一因素异常所造成的结果[7]。
从图1还可以看出,IL-18在乙肝引起的免疫反应中发挥了重要作用,一方面与IL-12一起诱导IFNγ生成,抑制病毒的复制,减轻感染;另一方面,IL-18还可诱导CD8+T细胞活化产生相应的细胞因子及产生细胞毒性作用,使感染细胞发生坏死。以前以为升高体内IL-18可以有利于病毒的抑制,但采用外源性的IL-18用于治疗乙肝引起的肝损伤并未获得成功。与此相反,后来的研究发现,乙肝患者血中的IL-18浓度显著高于正常人,且其浓度与肝病严重程度成正比[8]。有试验[9]发现,在多个肝纤维化模型大鼠的肝脏组织中,其IL-18浓度显著升高,而与此同时,IFNγ并未见升高,提示在慢性肝纤维化中,IL-18升高可能主要参与了诱导CD8+T活化使肝细胞坏死,其详细机理还有待进一步研究。
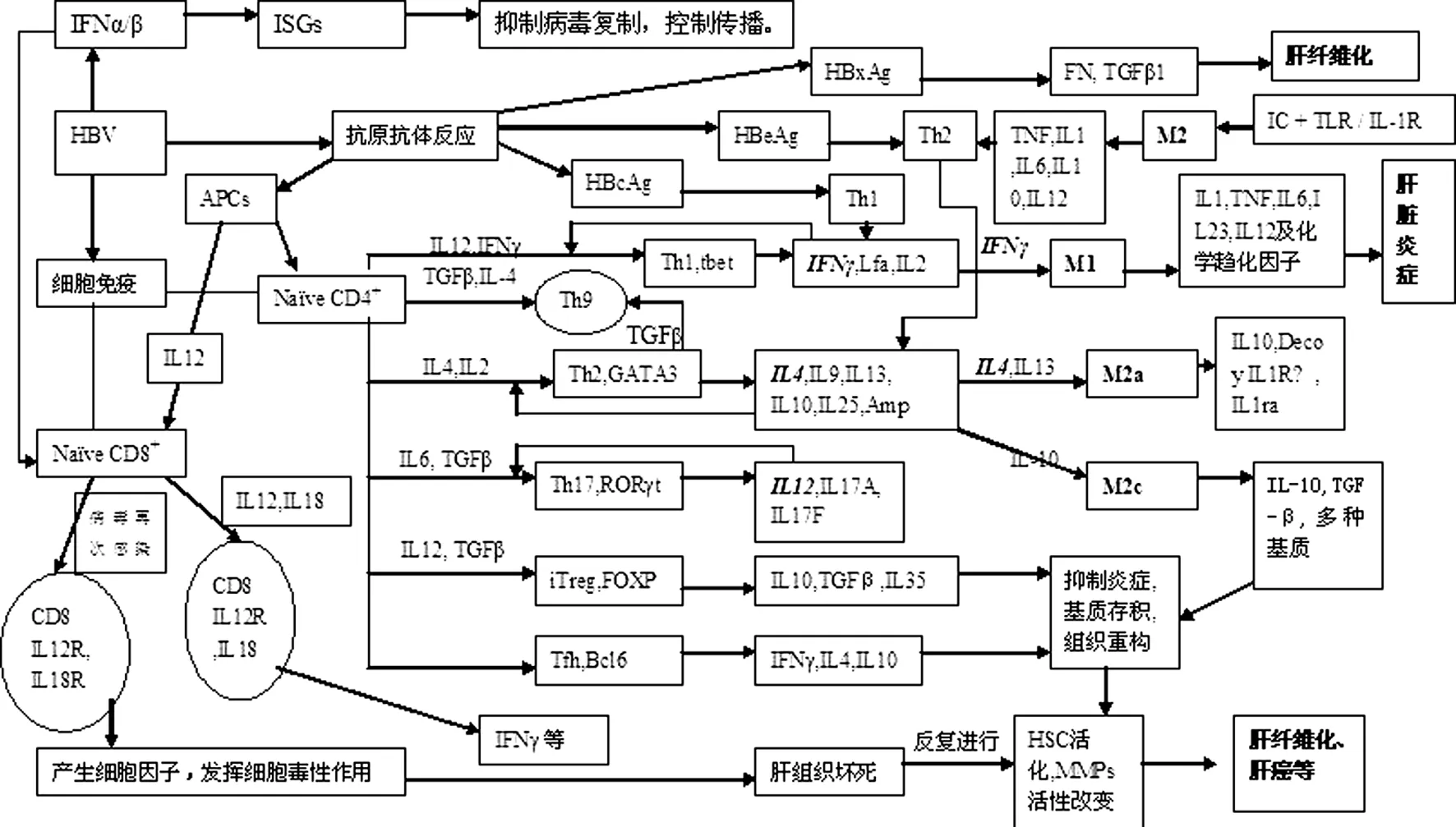
图1 乙肝病毒感染诱导肝纤维化发病机理示意图
图中英文简写的中文含义分别为:HBV(乙肝病毒),HBcAg(乙肝核心抗原),HBeAg(乙肝e抗原),HBxAg(肝细胞再生与HBVDNA碎片表达病毒克隆肝炎Bx抗原),NaïveCD4+(幼稚CD4+T细胞),NaïveCD8+(幼稚CD8+T细胞),iTreg(诱导性T调节细胞),Tfh(滤泡辅助性T细胞),M1、M2分别代表巨噬细胞两个亚型,IL(白介素),IFN(干扰素),R(受体),Th(辅助性T细胞)。HSC(肝星状细胞),TGF(转化生长因子)。
另外一个值得注意的细胞因子为IL-10。多个实验室[10,11]均发现多次注射ConA可引起IL-10 显著升高。从图1可见,IL-10在肝纤维化中的作用可能是通过诱导巨噬细胞M2c活化,产生TGFβ以及其它多种细胞外基质(ECM),促进肝纤维化。另外,IL-10 通过信号传递至T细胞使其增殖减少、效应子如IFNγ生成减少。IL-10可通过减少巨噬细胞和DCs的抗原呈递能力间接抑制T细胞应答,减少能够产生放大效应反应的促炎症细胞因子的表达[12]。
肝纤维化的特征是肝细胞减少、肝脏微小结构被破坏、肝脏肌纤维母细胞增殖以及细胞外基质过度堆积。终末期肝纤维化还可能包括肝脏解毒功能低下、肝细胞癌、门脉高压、肾功能以及肺脏功能衰竭,并导致死亡率显著增加。HBV引起肝纤维化的关键特征是纤维母细胞和肝星状细胞的增殖与活化。处于静息状态的肝星状细胞主要储存维生素A,存在于内皮下的狄氏间隙(spaceofDisse)。慢性肝损伤导致这些细胞活化、发生收缩、生成细胞外基质、分泌促炎性细胞因子和趋化因子,如转化生长因子β(TGFβ),这些细胞的活化被认为是肝纤维化的关键因素。肝星状细胞活化依赖于库否细胞(Kupffercells)、内皮细胞、肝细胞以及血小板的信号传递。细胞外基质的堆积与降解长期存在,随着肝纤维化的进程,该平衡向着有利于细胞外基质堆积倾斜。目前采用抗病毒治疗能够部分逆转肝纤维化的结果说明:肝纤维过程一直处于动态之中,由于病毒的长期存在,新的肝损伤时时都在发生,使肝纤维化逐步加重[13]。
目前西医防治乙肝所致的肝纤维化主要通过以下途径:a) 治疗原发病避免组织受损;b) 减轻炎症或宿主反应以避免刺激星状细胞活化;c) 保肝(hepatoprotection)以减轻肝脏损害,从而减轻刺激星状细胞活化的信号传递;d) 直接下调星状细胞活化;e) 抵消星状细胞增殖、纤维化、收缩和/或促炎症反应;f)刺激HSC凋亡;g) 增加疤痕组织降解,通过刺激细胞产生基质蛋白酶,或下调其抑制剂,或直接使用基质蛋白酶等[14]。
目前针对TGF-β信号通路的治疗策略如:(1)序列特异性反义寡核苷酸抑制TGF-βmRNA表达,(2)同型选择性中和抗体,可溶性TβRII碎片或合成的多肽干扰配体与内源性受体结合成复合体,(3)过表达自然TGF-β信号抑制剂Smad7或细胞因子如IFN-γ诱导Smad7表达,(4)中和整合素的抗体干扰潜TGF-β活化,(5)低分子量的抑制剂拮抗TGF-β受体的细胞内激酶的活性,这些策略在动物试验模型方面取得巨大成功,但是,这些策略用于临床的效果则微乎其微,导致这种结果的原因可能是TGF-β的功能具有生物多样性的特点,同时也说明肝纤维化并非TGFβ单一因素异常所造成的结果[8]
4 小 结
目前,对于肝纤维化的机理研究主要集中于肝纤维化的形成方面,近期的临床研究结果显示抗HBV药物对乙肝肝损伤有效,主要原因是其可以减少肝坏死和炎症,对于肝纤维化逆转的有效性有多高目前尚无准确的数据,但这些药物多存在一些不良反应;针对肝纤维化其它环节的药物疗效还需进一步验证。
近年来,随着对肝纤维化逆转机理的研究的深入,逐步注意到巨噬细胞在肝纤维化形成与降解中的重要作用,但要真正揭开肝纤维化形成与降解的全部机理还需进一步完成大量的工作。
