21羟化酶缺陷症32例临床分析
2012-12-17任高飞秦贵军刘会苗闫昱杉
任高飞,秦贵军,余 勤,刘会苗,李 俊,闫昱杉
郑州大学第一附属医院内分泌科郑州450052
#通讯作者,男,1965年8月生,教授,主任医师,研究方向:内分泌与代谢疾病,E-mail:hyqingj@zzu.edu.cn
先天性肾上腺皮质增生症(congenital adrenal hyperplasia,CAH)是由基因突变导致肾上腺皮质激素生物合成过程中必需酶缺陷从而使肾上腺皮质类固醇激素合成障碍引起的一组疾病[1],其中21 羟化酶缺陷症(21 hydroxylase deficiency,21-OHD)是最常见的类型,占90%~95%[2]。21-OHD 为常染色体隐性遗传病,是导致新生儿假两性畸形的常见病因,其发病率具有种族差异性[3]。21-OHD 包括经典失盐型、单纯男性化型和非经典型,其中前两者又统称经典型。作者回顾性分析了就诊的32例21-OHD 患者的临床特点。
1 临床资料
1.1 一般资料 2002年8月至2010年7月郑州大学第一附属医院内分泌科及小儿内科收治的32例21-OHD 患者,诊断依据临床表现和激素水平。社会性别男14例,女18例;其中1例社会性别为男性者染色体性别为女性,余社会性别均与染色体性别一致。
入院后检测各种血、尿相关激素水平。其中睾酮(T)、雌二醇(E2)、卵泡生成素(FSH)、黄体生成素(LH)及孕酮(P)采用酶免疫法测定;促肾上腺皮质激素(ACTH)、皮质醇(COR)采用电化学发光法测定;17 羟孕酮(17-OHP)、醛固酮(ALD)、尿游离皮质醇(UFC)采用放免法测定; 尿17-羟皮质类固醇(17-OH)、尿17-酮皮质类固醇(17-KS)采用化学法测定;血清电解质采用罗氏6400 速全自动生化分析仪测定。21例行16 排CT,11例行64 排CT。
1.2 临床表现和体征 32例患者年龄为3 d~22岁,发病年龄(3.8±5.7)岁,诊断年龄(8.5±7.8)岁。①经典失盐型13例,其中染色体性别男11例,女2例;1例3 岁,余12例均小于1 岁。5例有家族史,11例以呕吐、纳差为主诉,9例合并阴蒂或阴茎肥大,13例均合并低钠血症,11例合并高钾血症,10例合并低氯血症。②单纯男性化型19例,其中染色体性别男2例,女17例,1例女性社会性别为男性,年龄4~22 (13.8±5.6)岁,2例男性患者均存在第二性征提前出现,出现年龄(4.5±0.7)岁。女性患者中,15例以原发性闭经就诊,仅2例出现月经初潮,但极不规律;5例女性患者幼年时已行阴蒂切除并成形术,但均未给予病因诊断; 均伴有阴蒂肥大,并逐渐出现胡须旺盛、声音低沉、色素沉着、痤疮等男性化表现。
1.3 激素及影像学检查 经典失盐型患者年龄多在1 岁以内,激素测定及影像学检查相对不完善,故仅报告单纯男性化型患者的相关检查结果,见表1。由表1可知,血COR 及UFC 尚在正常范围,ACTH、血17-OHP、血T 及尿17-KS 平均水平均明显高于正常。促性腺激素水平均在正常范围。影像学检查示14/19 患者出现骨龄提前,16/19 患者有不同程度的双侧肾上腺增生。染色体性别为女性的1例患者妇科B 超检查子宫为幼稚型,6/17 患者卵巢呈多囊改变。
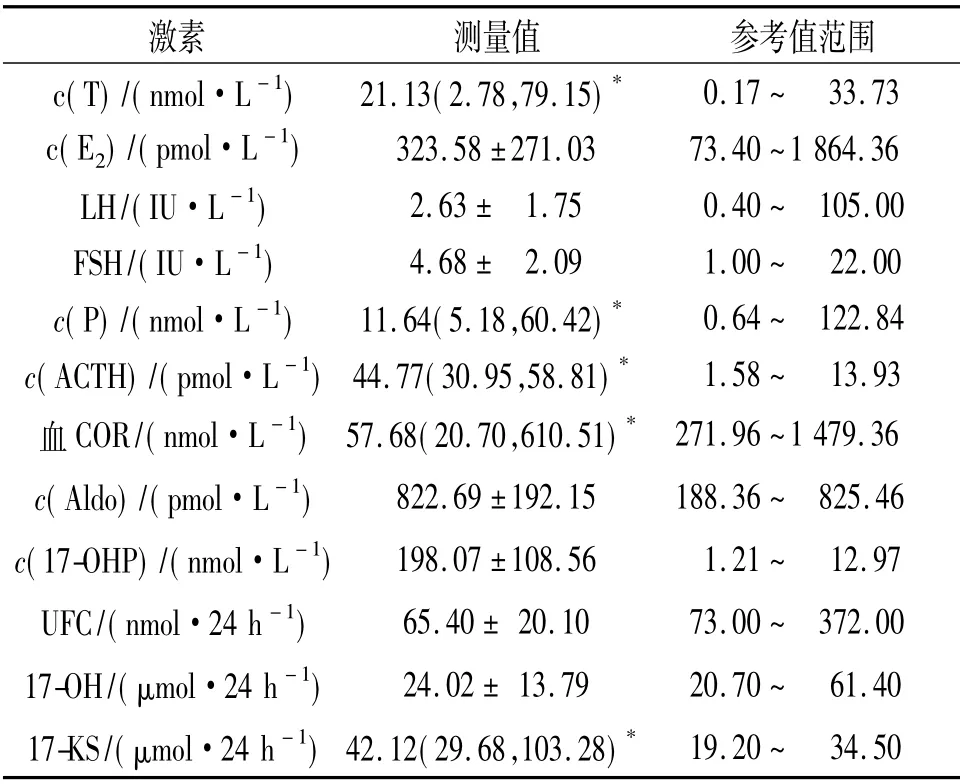
表1 单纯男性化型患者基础激素水平
2 讨论
21 羟化酶在肾上腺皮质激素的合成过程中起重要作用,该酶缺陷使得P 和17-OHP 不能羟化,导致盐皮质激素和糖皮质激素合成受累,从而引起ACTH 过度分泌,刺激肾上腺皮质增生和肾上腺源性雄激素合成和分泌增加。目前已发现100 多个基因突变点与21-OHD 有关,包括点突变、小片段丢失、基因重组等形式,故其临床表现也相差较大[4]。当21 羟化酶完全缺乏时,皮质激素合成绝对不足,即出现经典失盐型表现,此型通常出生时即可出现脱水、低钠及高钾性酸中毒,病死率极高。该组病例中有13例经典失盐型,仅1例于3 岁时发病,其余均于出生后不久出现纳差、呕吐、皮肤色暗等症状,所有患儿均合并低钠血症。单纯男性化型患者醛固酮合成正常,女性患者出现假两性畸形,男性患者出生时表型正常,于幼年出现性早熟、阴毛早现及生长加速等假性早熟症[5]。该组病例中单纯男性化型19例,2例男性均于幼年出现假性早熟症;17例女性患者存在典型男性化表现,其中1例社会性别为男性。非经典型21-OHD 因其症状隐匿,临床漏诊率高,阴毛早现可作为重要提示,患者于青春期或成年常合并多毛、痤疮、秃顶及多囊卵巢等[2]。
实际临床工作中21-OHD 的诊断除了临床表现外,很大程度上要依赖血17-OHP 水平。仅凭一次随机血17-OHP 水平超过242 nmol/L 就可以诊断[6]。但非典型的21-OHD 患者临床症状不典型且17-OHP 可正常,此时快速ACTH 兴奋实验在临床诊断上有重要意义。患者ACTH 兴奋实验后60 min 17-OHP 水平达到45.5 nmol/L 以上就可以诊断[1]。基因突变分析将更有助于明确诊断。该组患者血17-OHP 平均水平达(198.07±108.56)mmol/L,大大超出正常参考值。
Speiser 等[7]推荐对于生长发育阶段的21-OHD患者使用氢化可的松片剂,同时对于经典型21-OHD在新生儿期还应补充氟氢可的松和氯化钠;对于成年患者,地塞米松半衰期长,能更有效地抑制ACTH 的分泌;药物治疗期间建议进行激素水平监测。
总之,经该研究总结,经典失盐型21-OHD 患儿的临床变现以呕吐、纳差为主要表现,均合并低钠血症;单纯男性化型21-OHD 女性患者原发性闭经为就诊的主要原因,多合并多囊卵巢。血17-OHP 水平检测对诊断具有特异性。
[1]Miller WL.Molecular biology of steroid hormone synthesis[J].Endocr Rev,1988,9(3):295
[2]宁光,杨军.非经典型21-羟化酶缺陷症的诊治[J].中国实用内科杂志,2007,27(23):1816
[3]New MI.Nonclassical 21-hydroxylase deficiency[J].J Clin Endocrinol Metab,2006,91(11):4205
[4]Nimkarn S,New MI.Congenital adrenal hyperplasia due to 21-hydroxylase deficiency:a paradigm for prenatal diagnosis and treatment[J].Ann N Y Acad Sci,2010,1192:5
[5]Cabrera MS,Vogiatzi MG,New MI.Long term outcome in adult males with classic congenital adrenal hyperplasia[J].J Clin Endocrinol Metab,2001,86(7):3070
[6]New MI,Lorenzen F,Lerner AJ,et al.Genotyping steroid 21-hydroxylase deficiency:hormonal reference data[J].J Clin Endocrinol Metab,1983,57(2):320
[7]Speiser PW,Azziz R,Baskin LS,et al.Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency:an endocrine society clinical practice guideline[J].J Clin Endocrinol Metab,2010,95(9):4133
