药物非临床毒代动力学研究进展
2012-01-16杜武许家星张娟刘兆华刘兆平
杜武,许家星,张娟,刘兆华,刘兆平
(山东大学1.药学院,2.新药评价中心,山东济南250012)
药物非临床毒代动力学研究进展
杜武1,2,许家星2,张娟1,刘兆华2,刘兆平2
(山东大学1.药学院,2.新药评价中心,山东济南250012)
药物非临床毒代动力学是药代动力学在全身暴露评价中的延伸,或为非临床毒性研究的一部分,或为某一专设支持性研究,可为药物临床试验或应用提供安全性依据。目前毒代动力学已由最初的解释毒性试验结果逐渐扩展至毒性机制研究,成为在药物非临床和临床研究间的桥梁,同时其研究范围也扩展至药物代谢产物等的安全性评价中。本文就毒代动力学的实施、复合因素以及国内外研究进展进行综述,并展望其发展新方向。
药代动力学;毒性作用;评价研究
药物非临床毒代动力学是药代动力学在全身暴露评价中的延伸,其可结合于各种毒性试验而成为毒性研究的一部分,也可独立于毒性试验之外而成为专设支持性研究[1]。毒代动力学通过测定药物及其相关物质体内暴露的血浆或组织浓度,获得剂量与体内暴露水平之间的线性或非线性规律,用于阐明毒理学发现或毒性反应与剂量或血药浓度之间的关系,最终为药物临床试验或应用提供安全性依据。
由于现代分析方法的发展和应用,毒代动力学研究由最初的解释毒性实验结果逐渐扩展至毒性机制研究,并日益成为药物非临床研究的一项重要评价内容;同时其作为药物非临床和临床研究之间的桥梁[1],将两者更加紧密地联系在一起。此外,毒代动力学研究亦扩展至药物代谢产物和新型药物辅料的安全性评价中,为在实验动物体内评价可能暴露于人体的药物相关物质安全性研究提供了有力支持。本文就毒代动力学的实施、复合因素及在国内外研究进展进行综述。
1 毒代动力学研究及影响其实施的因素
1.1 毒代动力学研究的实施
毒代动力学研究通常结合于各种毒性试验中,被称为“伴随毒代动力学”,亦可在模拟毒性研究条件下的支持性实验中开展。伴随毒代动力学是药物非临床安全性评价的组成部分,其应严格遵循药物非临床研究质量指导规范进行实验;而在模拟毒性研究的条件下,回顾性设计并以获取安全性评价数据为目的的毒代动力学研究也应遵循非临床研究质量指导规范进行实验[1]。
毒代动力学研究一般需选定合适实验动物,以合理给药途径、频率和剂量水平进行给药,于合理采样时间点采集含有药物原型和(或)代谢物的血浆或血清等样品并进行测定,最终获取血药峰浓度(maximum concentration,cmax)、药时曲线下面积(areaundercurve,AUC)和半衰期等常用暴露(0531)88380319参数[1]。近年来,平均血浆药物浓度也被推荐至药物安全性研究中,其通过计算单位时间内的AUC,用于解释药物剂量-效应等关系[2]。
1.2 影响毒代动力学研究的复合因素
诸多复合因素会增加毒代动力学研究实施的复杂性(图1),为提高以毒代动力学信息为基础预测人体安全性的准确性,需考虑特殊药物的性质、最适动物的选择、待测物质的确定、检测方法的选用、人体特殊性以及毒代动力学及参数与毒理效应之间的相关性等因素。
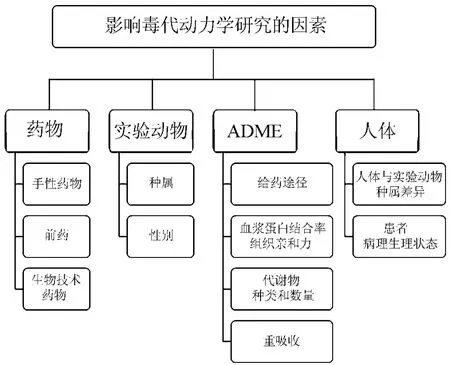
图1 影响毒代动力学研究的因素.ADME:机体对外源物的吸收、分布、代谢和排泄.
随着现代制药技术的进步,创新药物研发方兴未艾,这为毒代动力学研究提出了新的挑战。如手性药物,其同分异构体作用于同一靶点却可能具有不同亲和力,在采用气相色谱-质谱-单选择离子检测法,同时对丝氨酸对映体进行定量检测[3];如前药为修饰过的母药,其进入机体后被代谢为活性成分而发挥药效,因此,以前药的活性代谢物作为全身暴露的待测物质更为合理[4];肽类、蛋白类和单克隆抗体药物等生物技术药物,近年来也发展迅猛并成功用于临床治疗[5],在毒代动力学研究中,采用放射性同位素标记法、免疫印迹法以及ELISA法等新方法对相应药物进行检测[6-7]。
不同实验动物可能对同一药物显示不同灵敏度,如大鼠和犬在分别给予未见不良反应剂量水平(no observed adverse effect level,NOAEL)的止痛药物后,大鼠的血浆药物AUC值是犬的4倍,提示大鼠对此药物比犬更为敏感[8]。又如人源化的生物技术药物由于抗原性很难在常规实验动物体内实施检测,已有研究者尝试在黑猩猩和基因敲除小鼠上进行毒代动力学研究[9]。
药物在体内转运过程也存在诸多复合因素。药物吸收很大程度上取决于给药途径,给药途径的不同则有可能对毒代动力学检测产生影响;被吸收的药物可通过机体循环系统被转运至机体各器官组织,此时药物的血浆蛋白结合率及组织亲和力的异常,则会导致药物最终分布的异常。若药物可诱导或抑制代谢酶,则会导致药物及代谢物的异常累积或产生有毒代谢物而改变药物毒性作用。排泄过程中若存在肝肠循环、肾小管重吸收等现象,则会引起药物浓度波动进而干扰毒性研究结果。
人体与实验动物的种属间差异是预测药物人体安全性的首要障碍,如单克隆抗体TGN1412在实验动物中显示良好的安全性,但在临床试验中却出现严重不良反应[10];此外,患者人群多种病理生理状态也会影响预测的准确性,例如患者肝肾功能不全直接影响药物的清除,有极少数患者会对少量药物暴露产生过敏反应等。
2 毒代动力学的研究领域及意义
2.1 探索药物的剂量-暴露-毒性关系
传统毒性实验是通过剂量-毒性反应来研究受试药物的安全性,但对药物真正的体内暴露关注不足,而毒代动力学则可定量动态描述药物体内暴露情况,并通过毒代参数或药物浓度测量值为毒性实验结果解释提供依据。在重复给予受试药物后,实验动物体内药物暴露一般与剂量、暴露时间呈现相关性,如在柚皮苷(naringin)的毒代研究中发现,药物的暴露量与剂量水平增加呈比例关系[11];而药物暴露量的增加则极可能增加药物使用的风险性,如盐酸双苯氟嗪(dipfluzine)毒性与药物全身暴露呈线性[12]。此外,cmax和AUC值也可用于定量评价药物毒性,如在两种喹诺酮类药物的毒代动力学研究中,发现喹诺酮类药物900 mg·kg-1组的cmax和AUC0→24h值均低于氧氟沙星300 mg·kg-1阳性对照组的相应参数值,提示两种受试药物具有较低的毒性[13]。
2.2 寻找药物的毒性靶器官
机体某些组织器官可能由于大量血液灌流(如肝和肾)或对受试药物的高亲和力,引起过量药物及代谢物的积蓄继而受损,而毒代动力学的药物浓度测量值和各种参数,在揭示毒性靶器官和组织研究中发挥着重要作用。如在9-〔2-(膦酰甲氧基)乙基〕腺嘌呤一钠盐毒代研究中,肾清除率的下降提示药物对肾的毒性作用[14];在两性霉素B的毒代研究中,脾、肝和肾因蓄积高浓度的受试药物而被疑为毒性靶器官[15];司帕沙星在关节软骨中的浓度远高于血浆药物浓度,提示前者可能为药物毒性的靶组织[16]。
2.3 毒性生物标志物的发现
近年来,毒性生物标志物因在评价药物毒性损害时更为直观简便而广受关注,而毒代动力学在毒性生物标志物的研究中有着巨大潜力。在MLN8866的毒代研究中,血清淀粉样蛋白A和触珠蛋白的增长水平与受试药物的全身暴露(以cmax和AUC0-4d表征)和毒性结果成正相关关系,提示上述两种蛋白可作为MLN8866治疗时潜在的毒性生物标志物[17]。在特殊情况下,当药物毒性标志物不易获取时,传统监测手段依然可以与毒代动力学研究相结合达到毒性监测的目的。如在诺氟沙星的中枢神经毒性研究中发现脑电图的总响应值与药物AUC0→∞相关联,提示EEG可作为诺氟沙星中枢神经毒性早期诊断方法之一[18]。
2.4 非线性动力学研究
有些药物的体内过程(ADME)存在酶或载体的参与,当给药剂量及药物体内浓度达到一定限度时,酶催化或载体转运能力即达到饱和,使药代动力学参数随剂量改变而变化,从而出现非线性动力学现象。例如维生素B2通过近端小肠的非Na+依赖性载体以主动转运方式吸收[19],一定剂量的维生素B2可饱和该转运载体,使药物的吸收率不随剂量增加而变化[20]。药物毒性实验剂量设置是以假定药物的剂量与毒性呈正比关系为前提的,而药物的非线性动力学性质可能导致毒性研究中的剂量限制或使毒理学发现无效,此时毒代动力学研究将非常有助于评价剂量与暴露间的相关性[1]。如在两性霉素B脂质体的毒代研究中,发现药物的分布可被饱和,药物清除率和分布容积均随着剂量增加而下降,提示其具有非线性动力学特性;同时通过对药物毒性定量评估,发现药物毒性仍与剂量呈线性关系,证实药物毒性风险并未因药物的非线性动力学特性而增加[15]。此外,有些药物在一定剂量下可诱导或抑制代谢酶而使药物消除异常,亦可引起非线性动力学现象。如在阿伐麦布(avacimibe)的毒代动力学研究中比较了其在第1和第9天的cmax和AUC值,发现所有剂量组下该数值均降低,提示受试药物可能诱导肝CYP3A酶而出现非线性动力学现象[21]。
2.5 药物代谢产物研究
有些药物可被代谢酶代谢成为毒性产物,而其毒性代谢产物将加剧细胞损伤而出现器官毒性。以对乙酰氨基酚为例,大量药物被服用后会被肝药酶直接氧化生成乙酰苯醌亚胺,乙酰苯醌亚胺将破坏拓扑异构酶Ⅱα而最终引起肝细胞大量坏死[22]。同样,当受试药物被代谢为药理毒理活性产物并产生明显组织器官反应时,毒代动力学检测将非常有助于揭示药物毒性产生机制。如药物CB1954在临床试验中显示具有肝毒性,于是选用CD-1裸鼠和SD大鼠,分别给予以最大耐受量水平的药物并对其去硝基代谢产物进行测定,结果显示代谢物在小鼠肝中浓度高于血浆水平,而大鼠体内代谢物只存于肝而未见于血浆中,确证了药物毒性代谢物与肝毒性的关联性[23];又如新型抗焦虑药物PNU-101017在毒性试验中可引起实验动物死亡,通过检测药物的代谢产物对其毒性发生机制进行了深入探讨[24]。
此外由于种属间差异性,有些药物在人体内可能被代谢为非比例高水平代谢产物,后者需在实验动物中进行进一步的安全性研究。指导原则[25]规定,如果临床研究中发现的代谢产物未见于测试动物中,或其在测试动物中暴露水平远低于人体,则需进行进一步的动物试验以确定该代谢产物的潜在毒性;此时,应首先确认一种可形成该代谢产物充分暴露水平(与人体暴露量相当或更高)的动物种属,并于该动物种属中研究药物毒性;若不能确认相关动物种属,则可合成该代谢产物并直接给予动物以进一步进行安全性评估。因此,待测代谢产物在实验动物中的充分暴露是该研究的关键,而毒代动力学将非常有助于保证待测代谢产物的充分暴露,这为药物代谢产物安全性研究提供了有力支持[25]。
2.6 毒代动力学与临床研究
药物研发是贯穿于非临床和临床研究的动态反馈过程,毒代动力学作为其间的桥梁将两者更加紧密地联系在一起。毒代动力学测定的血/组织药物浓度是药效/毒效的物质基础,如果非临床研究可以发现或找到血/组织药物浓度与毒效或无毒性反应之间的关系,则可以应用血药浓度这一桥梁在人体试验中通过药物剂量-浓度关系来保证临床安全性。如新药Ⅰ期临床试验以最大推荐起始剂量作为人体首剂最大安全起始剂量,最大推荐起始剂量首先以非临床动物毒性试验的NOAEL剂量换算为人体等效剂量,然后选择最低人体等效剂量除以安全系数(一般为10)而得[26]。毒代动力学可对实验动物NOAEL水平下药物暴露进行监测,并与人体最大推荐起始剂量下的药物暴露量进行比较,从而辅助确认最大推荐起始剂量的合理性并保障受试者的安全[8]。
此外,在实验动物中回顾性地开展毒代动力学试验,可对药物临床试验信息进行验证和深入研究。如CB1954在临床试验中显示具有肝毒性,研究者通过开展毒代动力学试验,最终确认毒性代谢物与肝毒性的关联性[23];而于代谢产物的安全性研究中开展毒代动力学研究,对保证受试物充分暴露起到关键作用[25]。
2.7 其他
药物经不同给药途径进入机体可能会具有不同药物暴露情况,故拟改变给药途径的药物需考虑进行毒代动力学评价;另外已上市药物合用并作为复方药物开发时,由于药物的动力学性质可能发生改变,亦需毒代动力学对药物暴露情况进行评价[8];而新型药物辅料近年来被认定为新化学实体,必要时也需进行毒代动力学的评价[27-28]。
3 展望
高效液相色谱-质谱和高效液相色谱-质谱-质谱等仪器分析方法为毒代动力学检测提供了坚实的基础;也正是由于分析方法的进步,更加深入地寻找与检测毒性代谢产物逐渐被纳入毒代动力学研究领域中,这将有利于阐明药物毒性发生机制并完善药物安全性研究。同时新药研发是贯穿于非临床和临床研究的动态反馈过程,毒代动力学作为其间的桥梁将两者更加紧密地联系在一起,而其在药物代谢产物等药物相关物质的安全性研究中的作用也将愈加重要。总之,毒代动力学日益成为药物非临床研究的一项重要评价内容,相信随着创新药物研发的不断深入,其必将发挥更大的作用。
[1]FDA.Toxicokinetics:The Assessment of Systemic Exposure in Toxicity Studies[EB/OL][1995-05-25]http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM074937.pdf
[2]Smith DA,Morgan P,Vogel WM,Walker DK.The use of C(av)rather than AUC in safety assessment[J].Regul Toxicol Pharmacol,2010,57(1):70-73.
[3]Hasegawa H,Shinohara Y,Masuda N,Hashimoto T,Ichida K.Simultaneous determination of serine enantiomers in plasma using Mosher's reagent and stable isotope dilution gas chromatographymass spectrometry[J].J Mass Spectrom,2011,46(5):502-527.
[4]Ravel D,Dubois V,Quinonero J,Meyer-Losic F,Delord J,Rochaix P,et al.Preclinical toxicity,toxicokinetics,and antitumoral efficacy studies of DTS-201,a tumor-selective peptidic prodrug of doxorubicin[J].Clin Cancer Res,2008,14(4):1258-1265.
[5]Lin JH.Pharmacokinetics of biotech drugs:peptides,proteins and monoclonal antibodies[J].Curr Drug Metab,2009,10(7):661-691.
[6]Coowanitwong I,Keay SK,Natarajan K,Garimella TS,Mason CW,Grkovic D,et al.Toxicokinetic study of recombinant human heparinbinding epidermal growth factor-like growth factor(rhHB-EGF)in female Sprague Dawley rats[J].Pharm Res,2008,25(3):542-550.
[7]Nordstrom JL,Gorlatov S,Zhang W,Yang Y,Huang L,Burke S,et al.Anti-tumor activity and toxicokinetics analysis of MGAH22,an anti-HER2 monoclonal antibody with enhanced Fcγ receptor binding properties[J].Breast Cancer Res,2011,13(6):R123.
[8]Baldrick P.Toxicokinetics in preclinical evaluation[J].Drug Discov Today,2003,8(3):127-133.
[9]Clarke J,Leach W,Pippig S,Joshi A,Wu B,House R,et al.Evaluation of a surrogate antibody for preclinical safety testing of an anti-CD11a monoclonal antibody[J].Regul Toxicol Pharmacol,2004,40(3):219-226.
[10]Stebbings R,Findlay L,Edwards C,Eastwood D,Bird C,North D,et al."Cytokine storm"in the phaseⅠtrial of monoclonal antibody TGN1412:better understanding the causes to improve preclinical testing of immunotherapeutics[J].J Immunol,2007,179(5):3325-3331.
[11]Liu M,Yang C,Zou W,Guan X,Zheng W,Lai L,et al.Toxicokinetics of naringin,a putative antitussive,after 184-day repeated oral administration in rats[J].Environ Toxicol Pharmacol,2011,31(3):485-489.
[12]Hu H,Wang Y,Pei T,Dong L,Xu Y.Acute toxicity and toxicokinetics of dipfluzine hydrochloride,a novel calcium channel blocker[J].Regul Toxicol Pharmacol,2009,54(1):66-71.
[13]Goto K,Yabe K,Suzuki T,Jindo T,Sanbuissho A.Chondrotoxicity and toxicokinetics of novel quinolone antibacterial agents DC-159a and DX-619 in juvenile rats[J].Toxicology,2010,276(2):122-127.
[14]Wang WY,Shen ZL,Yao QS,Yao J,Bai WX,Pan YY.Toxicokinetics and toxicological studies of sodium 9-[2-(phosphonomethoxy)ethyl]adenine in beagle dogs[J].Chin J Pharm Toxicol(中国药理学与毒理学杂志),2006,20(6):461-467.
[15]Bekersky I,Boswell GW,Hiles R,Fielding RM,Buell D,Walsh TJ.Safety and toxicokinetics of intravenous liposomal amphotericin B(AmBisome)in beagle dogs[J].Pharm Res,1999,16(11):1694-1701.
[16]Stahlmann R,Zippel U,Förster C,Schwabe R,Shakibaei M,Merker HJ,et al.Chondrotoxicity and toxicokinetics of sparfloxacin in juvenile rats[J].Antimicrob Agents Chemother,1998,42(6):1470-1475.
[17]Hsieh FY,Tengstrand E,Li LY,Huang YN,Milton MN,Silverman L,et al.Toxicological protein biomarker analysis-an investigative one-week single dose intravenous infusion toxicity and tox-icokinetic study in cynomolgus monkeys using an antibody-cytotoxic conjugate against ovarian cancer[J].Pharm Res,2008,25(6):1309-1317.
[18]Zhang LR,Wang YM,Chen BY,Cheng NN.Neurotoxicity and toxicokinetics of norfloxacin in conscious rats[J].Acta Pharmacol Sin,2003,24(6):605-609.
[19]Said HM.Recent advances in carrier-mediated intestinal absorption of water-soluble vitamins[J].Annu Rev Physiol,2004,66:419-446.
[20]Guo L,Lin DS.Brief discussion about improving the bioavailability of oral medicine[J].Heilongjiang Med J(黑龙江医学),2005,29(9):096.
[21]Robertson DG,Breider MA,Milad MA.Preclinical safety evaluation of avasimibe in beagle dogs:an ACAT inhibitor with minimal adrenal effects[J].Toxicol Sci,2001,59(2):324-334.
[22]Bender RP,Lindsey RH Jr,Burden DA,Osheroff N.N-acetyl-pbenzoquinone imine,the toxic metabolite of acetaminophen,is a topoisomeraseⅡpoison[J].Biochemistry,2004,43(12):3731-3739.
[23]Tang MH,Helsby NA,Goldthorpe MA,Thompson KM,Al-Ali S,Tingle MD.Hepatic nitroreduction,toxicity and toxicokinetics of the anti-tumour prodrug CB 1954 in mouse and rat[J].Toxicology,2007,240(1-2):70-85.
[24]Zhong WZ,Williams MG,Branstetter DG.Toxicokinetics in drug development:an overview of toxicokinetic application in the development of PNU-101017,an anxiolytic drug candidate[J].Curr Drug Metab,2000,1(3):243-254.
[25]FDA.Safety Testing of Drug Metabolites[EB/OL][2008-10-17]http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM079266.pdf
[26]FDA.Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers[EB/OL][2005-08-12]http://www.fda.gov/downloads/Drugs/Guidance-ComplianceRegulatoryInformation/Guidances/UCM078932.pdf
[27]Baldrick P.Pharmaceutical excipient development:the need for preclinical guidance[J].Regul Toxicol Pharmacol,2000,32(2):210-218.
[28]Li BQ,Dong X,Fang SH,Gao JY,Yang GQ,Zhao H.Systemic toxicity and toxicokinetics of a high dose of polyethylene glycol 400 in dogs following intravenous injection[J].Drug Chem Toxicol,2011,34(2):208-212.
Progress in nonclinical toxicokinetics in drugs
DU Wu1,2,XU Jia-xing2,ZHANG Juan1,LIU Zhao-hua2,LIU Zhao-ping2
(1.School of Pharmacy,Shandong University,Jinan250012,China;2.Center for New Drug Evaluation of Shandong University,Jinan250012,China)
Toxicokinetics is normally performed as an integral component in nonclinical toxicity studies or as a part in specially designed supportive studies,and it can generate kinetic data at different dose levels for assessment of systemic exposure and providing scientific support evidences for clinical safety issues.Toxicokinetic study has been gradually extended from only explaining toxicity test results to investigate toxic mechanism.It acts as a bridge between nonclinical and clinical studies regarding the drug safety,and becomes more crucial than ever before.Its scope has been further expanded to safety evaluation of drug related substances,such as drug metabolites.This paper reviewed the test procedures and complicating factors of toxicokinetic study,and its roles in new drug development,and a prospect of the further development is also summarized.
pharmacokinetics;toxic actions;evaluation studies
The project supported by National Science and Technology Major Projects(2009ZX09502-001);and National Natural Science Foundation of China(30973934)
LIU Zhao-ping,E-mail:liuzhaoping@sdu.edu.cn,Tel:(0531)88380319
R969.1
A
1000-3002(2012)06-0903-04
10.3867/j.issn.1000-3002.2012.06.022
国家科技重大专项课题(2009ZX09502-001);国家自然科学基金(30973934)
杜武(1986-),男,硕士研究生,主要从事药物毒理学研究;刘兆平(1958-),男,教授,硕士生导师,主要从事药物安全性评价研究。
刘兆平,E-mail:liuzhaoping@sdu.edu.cn,Tel:
2011-11-17接受日期:2012-03-21)
(本文编辑:付良青)
