无菌检查中污染微生物多样性分析及数据偏差调查*
2024-05-07王璐,向东,张艳,刘勤,徐帆
王 璐,向 东,张 艳,刘 勤,徐 帆
(1. 云南省食品药品监督检验研究院·工业和信息化部产业技术基础公共服务平台,云南 昆明 650106;2. 中国人民解放军联勤保障部队第九二〇医院,云南 昆明 650032)
无菌检查法用于检查药典规定无菌的药品、生物制品、医疗器具、原料、辅料及其他品种是否无菌,是注射用药品安全性评价指标之一。无菌检查结果的判断是一个非常重要和复杂的步骤。国际人用药品注册技术协调会(ICH)规定,无菌检出结果以一次检出为准,不得复试[1]。2020年版《中国药典》规定,当供试品管检出微生物时,若有证据能充分证明生长的微生物非供试品本身引起,可判定试验无效,可重试,否则应判定供试品不符合无菌检查法的要求[2]。无菌检查判断如出现假阴性结果,会造成已污染微生物的无菌药品流向市场(如“欣弗事件”“刺五加事件”),严重威胁患者生命;如出现假阳性结果,因无菌药品生产企业均为高风险产品企业,企业必须召回上市产品,停业整顿,给企业带来不可估量的损失,同时也会对药品监管检验机构的公信力造成不良影响。药品生产企业或药品检验机构应具备微生物污染溯源的能力。首先应对污染微生物进行分离纯化,根据染色镜检结果采用生化反应鉴定法确定细菌种属,并与环境微生物数据库进行比对。当日常微生物或生化鉴定技术灵敏度偏低或不足以提供明确的证据证明2 种分离的微生物为同一来源时,应采用更敏感的试验方法(如基因型微生物鉴定)来测定分离的微生物是否为克隆关系及由同一菌株繁殖而成。无菌检查法作为终产品检查法虽能防止问题产品流向消费领域,却无法对药品检验进行过程控制,无法解决药品微生物污染来源问题。本研究中以曲安奈德注射液为例,旨在探索当药品的无菌检查出现阳性结果后,样品微生物与环境微生物之间的类聚特征与同源性关系,并结合本实验室的微生物数据偏差数据调查(MDD)程序,以期排查从样品到检验过程的污染原因,追溯阳性微生物的来源。现报道如下。
1 仪器与试药
1.1 仪器
STI- 1800DTC 型无菌隔离器(浙江泰林生物技术股份有限公司);HPP260eco 型生化培养箱(德国Memmert 公司);AC2 - 4S1 级A2 型生物安全柜(ESCO Airstream®,新加坡ESCO 公司);DM300 型LED 正置显微成像系统(德国Leica公司);VITEK 2 Compact型全自动细菌鉴定及药敏分析系统(法国Bio Mérieux公司)。
1.2 试药
曲安奈德注射液(国内A 企业,批号为220724,规格为每支1 mL);硫乙醇酸盐流体培养基(FTM)及胰酪大豆胨琼脂、液体培养基(TSA,TSB),均购自北京三药科技开发公司,批号分别为20220405,220331,20220412;革兰阴性细菌及革兰阳性细菌鉴定卡(GP 及GN,美国Bio Mérieux 公司,批号分别为2411814203,2422281503)。
2 方法与结果
2.1 无菌检查
曲安奈德注射液为混悬液无菌产品,按2020年版《中国药典(四部)》通则1101无菌检查法下直接接种法进行无菌检查。培养期间定期观察接种了供试品的FTM,第10 天发现其中1 管氧化层颜色变淡,因制剂本身为白色混悬液,接种后培养基管均出现浑浊,故从外观上不能判断有无微生物生长。培养至第14天,取该培养液1 mL 置同种新鲜培养基中,继续培养不少于4 d,新鲜培养基再出现浑浊,则判断为有菌生长。
2.2 细菌的分离纯化及鉴定
2.2.1 细菌的分离培养及染色
取上述培养物,用TSA 划板,分离纯化,培养结果染色镜检[3],结果见表1。

表1 镜检及生化鉴定结果Tab.1 Results of microscopic examination and biochemical identification
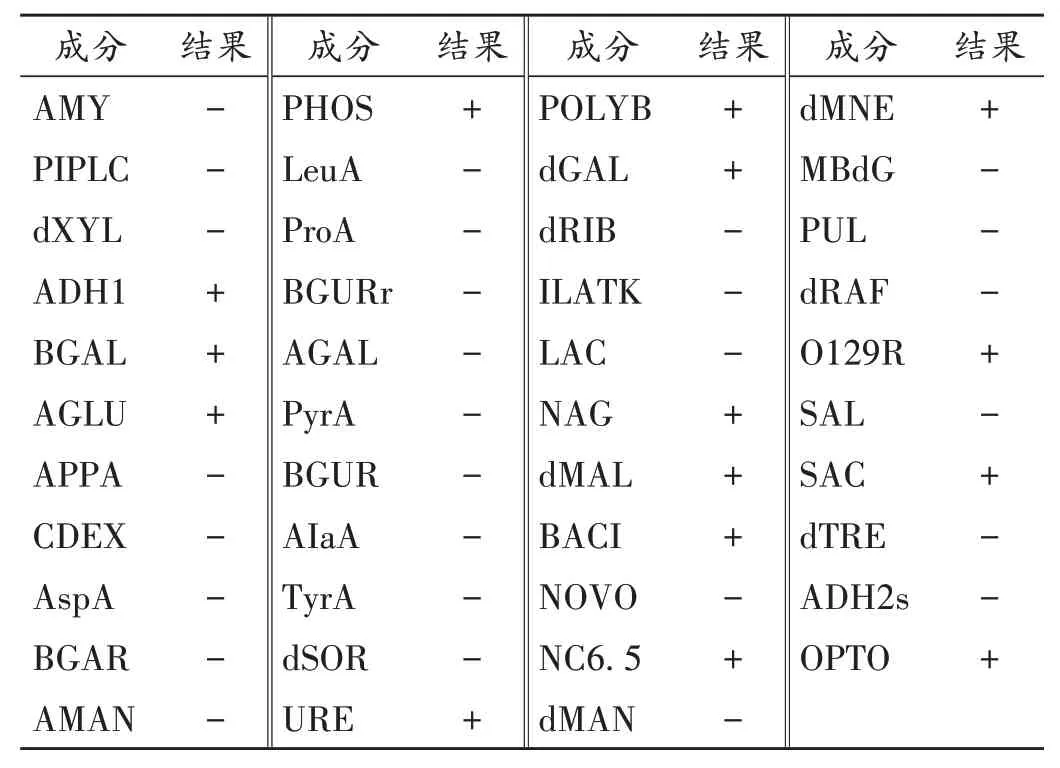
表2 表皮葡萄球菌生化鉴定结果Fig.2 Results of biochemical identification of Staphylococcus epidermidis
2.2.2 全自动细菌生化鉴定
根据革兰染色镜检结果,选择对应的生化鉴定卡片,对分离纯化得到的2 个菌株进行生化鉴定,试验结果置信度极佳。详见表1 至表3(其中,+ 代表阳性,-代表阴性)。
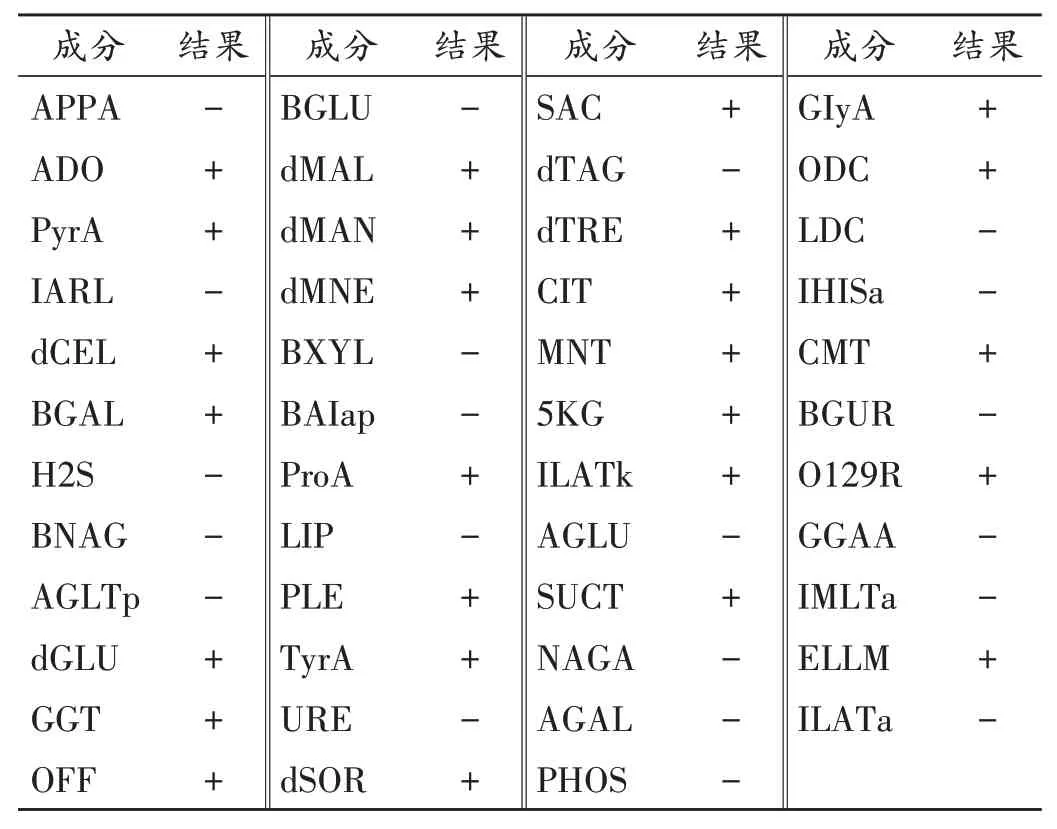
表3 克氏柠檬酸杆菌生化鉴定结果Fig.3 Results of biochemical identification of Citrobacter koseri
2.2.3 微生物多样性分析
委托北京擎科生物科技股份有限公司昆明分公司对出现浑浊长菌的培养管进行微生物多样性分析。提取样品总DNA 后,根据DNA 保守区设计引物(在引物末端加上测序接头)进行聚合酶链反应(PCR)。并对其产物进行纯化、定量和均一化,形成测序文库,对原始测序序列进行质量控制(包括低质量过滤、长度过滤),得到高质量序列,参数内容见表4。

表4 参数内容与信息Tab.4 Content and information of parameters
对高质量序列进行过滤及去噪,首先使用Trimmomatic v0.33软件对测序得到的Raw Reads 进行过滤;然后使用cutadapt 1.9.1 软件进行引物序列的识别与去除,得到不包含引物序列的Clean Reads。第二步,DADA2去噪:使用QIIME2 2020.6[4]中的DADA2 方法[5]进行去噪,双端序列拼接并去除嵌合体序列,得到最终有效数据(Non-chimeric Reads)。
信息分析内容,划分OTUs/ASVs(Feature),并根据Feature 的序列组成得到其物种分类。进一步进行多样性分析、差异分析、相关性分析及功能预测分析。最后根据16S rRNA 或内转录间隔区(ITS)基因测序结果,通过功能预测分析对样品进行基因功能或表型预测并计算功能基因或表型丰度,结果检测到3 种细菌,分别为表皮葡萄球菌、克氏柠檬酸杆菌及唾液链球菌。详见表5(其中,系列数指该微生物被检测到的系列条数,不代表该微生物的绝对数量;相对丰度指该微生物所占同类微生物的百分比)。

表5 16S rRNA系列分析结果Tab.5 Results of 16S rRNA analysis
2.3 与实验室环境菌库比较
收集2021年至2022年隔离器内部A级环境及隔离器放置的D 级背景的微生物,得39株环境菌。首先进行菌落形态特征观察,然后进行革兰染色,再用正置显微成像系统镜检,根据染色结果使用VITEK 2 Compact全自动微生物鉴定仪进行鉴定,对其中有疑问的进行16S rRNA 测序鉴定[6],结果包含了样品中检出的表皮葡萄球菌、克氏柠檬酸杆菌及唾液链球菌,具体为表皮葡萄球菌7 株,沃氏葡萄球菌6 株,克氏库克菌5 株,头状葡萄球菌3株,克氏柠檬酸杆菌、藤黄微球菌、唾液链球菌、肺炎链球菌各2株,腐生葡萄球菌、痤疮丙酸杆菌各1株,未鉴定出的有8株。
2.4 微生物数据MDD 分析[7]
按2020年版《中国药典(四部)》通则9203 药品微生物实验室质量管理指导原则,对检验过程中出现的与微生物相关的不合规范的数据进行实验室MDD。
仪器状态调查:隔离器系统安全舱灭菌程序调查,因当日实验样品较多,操作人员未按隔离器典型装载方式进行物料装载,目测物品装载已超过该台隔离器年度验证时的最大装载方式,部分样品装载时未完全分开,样品之间未充分暴露表面,因隔离器灭菌方式为气化过氧化氢表面灭菌,未暴露的表面可能因未接触到过氧化氢而不能充分灭菌。仪器本次和上次运行状态正常,日常维护与保养记录正常,仪器处于再验证合格期内。
培养基及冲洗液质量调查:培养基来源清楚、稳定,处于有效期内,保存条件满足说明书要求,其他试剂及耗材满足试验标准要求。
洁净保障:试验人员遵守无菌操作,手套检漏合格,沉降菌和表面微生物符合规定,本试验不涉及浮游菌监测,阴性对照及阳性对照符合规定。
检验操作过程:供试品的内外包装和取样量符合标准要求,检验标准使用正确。
其他需要说明的情况:当天同时进行的其他2批曲安奈德注射液无菌检查符合规定,未见异常。
纠正和预防措施:对所有无菌试验人员再次进行无菌隔离器使用规范培训,再次确认隔离器的典型装载方式及满载状态,确保今后试验中装载量不会超过经验证合格的仪器满载状态。此外,MDD 调查结果表明,样品中污染的微生物是由试验操作人员未完全按无菌隔离器使用的标准操作规程进行规范操作而引起的污染,污染菌来源于环境中。由于样品的装载方式存在问题,造成样品表面灭菌不完全。该样品的无菌检查法为直接接种法,导致培养管更易污染环境中的细菌。故本次无菌试验结果无效,应重试。
2.5 无菌试验重试结果
取同批次同量供试品,同2.1 项下方法进行无菌试验。将上述接种供试品后的培养基容器分别按各培养基规定的温度培养不少于14 d。培养期间逐日观察,结果阳性对照菌24 h 生长良好,阴性对照各管在培养期间无菌生长,供试品管无菌生长,判定样品无菌检查符合规定。
3 讨论
2020年版《中国药典(四部)》通则1101 规定,当无菌检查培养基任何1管浑浊并确认微生物污染时,若能证明污染的微生物非样品所含,可判定试验结果无效,并对该样品的无菌试验进行重试。对供试品管中有菌生长的无菌试验结果应认真分析产生的原因,如果分析原因符合下列其中1 条,可判定试验结果无效。1)无菌检查试验所用设备及环境的微生物监控结果不符合无菌检查法中对设备及环境的要求;2)回顾整个无菌试验操作过程,有充分证据表明在其中某一环节有可能引起供试品管的微生物污染;3)阴性对照管有微生物污染,当供试品管有菌生长时,阴性对照试验对判断无菌试验结果的有效性非常重要。供试品管中生长的微生物经鉴定后,应与环境微生物数据库中的数据进行比较,以确认是否为同一来源。但该判断方法与所用鉴定技术关系很大,需采用更敏感的试验来判断2种分离的微生物是否为克隆关系或由同一菌株繁殖而成,目前微生物常用鉴定同源性分析方法有生化反应鉴定法、16S rRNA 的微生物多样性分析、核糖体分型RP 鉴定方法、飞行时间质谱法、脉冲场凝胶电泳法等[8]。
微生物多样性研究是以扩增序列变体为分子标记研究环境样品中微生物系统分类、物种构成。16S rRNA为核糖体小亚基的组分,普遍存在于所有细菌中,具有拷贝数多、分子大小适中、有突变序列也有保守序列、信息量较大且能体现不同菌株种属间差异的特点,是细菌系统分类研究中最常用的“分子钟”,作为细菌系统发育和分类鉴定最常用的标准标识序列,素有“细菌化石”之称[9-10]。微生物多样性是基于Illumina Nova-Seq 测序平台,利用双末端测序(Paired - End)的方法,构建小片段文库进行测序。通过对Reads 拼接过滤,聚类或去噪,并进行物种注释及丰度分析,可揭示样品的物种构成;进一步进行α 多样性分析(Alpha Diversity)、β多样性分析(Beta Diversity)、显著物种差异分析、相关性分析、功能预测分析等,以挖掘样品之间的差异。目前,基于16S rRNA 的微生物多样性分析已广泛应用于菌株的鉴定研究。
目前,微生物污染的溯源依然是很多药品生产企业操作的难点,尽管如此,药品生产企业或药品检验机构仍应积极建立洁净区环境微生物数据库,企业可通过数据库实现对生产全过程的微生物监控,有利于生产各环节的控制,发现生产过程中的风险点,从被动的排查污染上升到主动预防,当监测数据超过警戒线和纠偏限时,就应高度重视,启动相应程序,让洁净区始终保持受控状态[11]。检验机构建立污染趋势分析图,可应用到溯源分析及污染调查中[12],当样品的检验结果异常时,调取环境中微生物数据库的菌种进行比对分析。因此,环境微生物数据库的建立对药品生产企业和药品检验机构来说均非常重要且必要。
