运动促进线粒体功能在改善神经退行性疾病中的分子机制
2024-02-21赵仁清朱柏树王元心周亚兰林俊杰
赵 晨,赵仁清,王 斌,朱柏树,王元心,周亚兰,林俊杰
神经退行性疾病 (Neurodegenerative Disease,NDD)主要是由神经元结构或功能丧失导致[1],它成为影响老年人健康的重要疾病。最近研究发现,大脑线粒体功能障碍是神经退行性疾病最显著的早期特征。在许多神经退行性疾病中均出现线粒体破碎、分布改变,表明异常的线粒体融合、裂变和运输可能导致线粒体功能障碍和神经退行性变。 运动可促进神经突触可塑性,增加记忆功能,在预防阿尔茨海默病(Alzheimer’s Disease, AD)、 帕金森病(Parkinson’s Disease, PD)等疾病中发挥有益作用。 由于线粒体功能障碍与神经退行性病变密切相关, 且运动可促进线粒体重塑,提高线粒体代谢功能,维持线粒体融合和分裂平衡,增加线粒体数量,因此,运动可能通过促进线粒体功能从而改善神经退行性疾病症状,然而,运动改善线粒体功能的分子机制复杂,且多种信号通路参与线粒体与神经退行性疾病病理过程。 从分子信号通路角度探讨调节运动促进线粒体功能、改善神经退行性疾病的机制,通过PubMed、Cochran、Embase、Web of Science、CNKI 及万方数据等数据库检索相关文献,检索时间由数据库建成至2023 年4 月,检索中文关键词 “神经退行性疾病”“线粒体功能障碍”“运动”,英文关键词“neurodegenerative diseases”“mitochondrialdysfunction”“exercise”,共纳入51 篇文献。
1 线粒体功能障碍与神经退行性疾病病理变化
线粒体功能障碍在神经退行疾病的病理过程中起关键作用, 任何线粒体功能障碍都会导致三磷酸腺苷(Adenosine Triphosphate, ATP)缺乏和神经元死亡(图1)。 许多神经退行性疾病都存在线粒体质量控制障碍, 线粒体自噬异常会导致线粒体碎片无法通过线粒体自噬被清除。此外,神经退行性疾病还出现其他显著变化,包括钙离子流入障碍、线粒体膜电位消失、线粒体突变蛋白的积累、线粒体DNA(Mitochondrial DNA, mtDNA) 缺乏以及线粒体氧化磷酸化(Oxidative Phosphorylation, OXPHOS)下降[2]。 在神经退行性疾病患者的神经细胞中出现线粒体合成ATP 减少、活性氧产生增加、钙缓冲能力下降,以及线粒体通透性增加[3]。

图1 线粒体功能障碍与神经退行性疾病病理生理变化Figure1 Mitochondrial dysfunction and pathophysiological changes of neurodegenerative diseases
1.1 线粒体融合和裂变
线粒体融合和裂变不平衡会导致其功能变化,包括脂质过氧化增加、 活性氧 (Reactive Oxygen Species, ROS)产生增加、膜电位下降、氧化呼吸降低,ATP 产生减少。 神经元对线粒体变化非常敏感,几乎所有调节线粒体融合和裂变的因子[如线粒体分裂基因动态相关蛋白1(Mitochondrial Dynamin-Related Protein 1, Drp1)、 视神经萎缩蛋白1(Optic Atrophy Protein 1, OPA1)、线粒体融合蛋白-2(Mitochondrial Fusion Protein 2, Mfn2) 和线粒体动力相关蛋白1(Mitochondrial Fission Protein 1, Fis1)]缺乏以及相关调节蛋白的异常表达都会损害线粒体的转运和形态,从而导致神经元突触中线粒体减少,最终出现突触和树突棘消失[4]。 有研究表明,线粒体的氧化磷酸化过程被抑制后,Drp1 表达降低以及膜去极化导致线粒体碎片增加[5],线粒体功能受损以及细胞死亡增加,最终导致神经元变性。与神经退行性疾病相关的蛋白质,如p-Tau、β- 淀粉样蛋白(Amyloid β,Aβ)、 突变亨廷顿蛋白 (Mutated Huntingtin Protein,mHtt)等,均可影响线粒体融合与裂变平衡,从而导致线粒体功能障碍, 最终导致神经元功能受损或死亡,诱发神经退行性疾病[6-7]。 因此,线粒体中融合和裂变之间的不平衡导致线粒体融合减少、 线粒体碎片化, 这是神经退行性疾病中细胞死亡和线粒体功能障碍的重要因素。
1.2 线粒体生物合成
线粒体合成是通过调节线粒体数量以适应细胞生长、 分裂和氧化应激与激素分泌变化而产生的能量需求。 线粒体生物合成缺陷会产生大量的ROS,以及造成mtDNA 损伤和蛋白质、脂质氧化障碍。 此外,ROS 还会通过线粒体通透性转运孔(Mitochondrial Permeability Transition Pore, mPTP)复合物蛋白质触发细胞内凋亡反应[8]。 线粒体生物合成过程主要受过氧化物酶体增殖物受体γ 共激活因子-1α(Peroxisome-Proliferator-Activated Receptor-γ-Coactivator-1α,PGC-1α)调节,调控翻译和复制mtDNA 的基因表达[9]。PGC-1α 在小脑神经元中过度表达可使线粒体密度增加30%, 而缺乏PGC-1α 的小鼠出现神经元缺失。PGC-1α 表达变化是神经退行性疾病的共同特征[10-11]。PGC-1α、核因子E2 相关因子(Nuclear Factor E2-Related Factor 2, Nrf2) 或线粒体转录因子A(Mitochondrial Transcription Factor A, TFAM) 在AD 患者的细胞核、脑组织中表达明显减少[12-13];PD 患者大脑中的PGC-1α 水平也明显降低[14];在亨廷顿病(Huntington’s Disease, HD)小鼠模型和患者纹状体中,PGC-1α mRNA 和蛋白质水平均降低[15]。 目前,在神经退行性疾病中发现线粒体生物合成变化与疾病密切相关,出现线粒体生物合成减少、出现功能障碍等变化。
1.3 线粒体ROS 与氧化应激
ROS 是线粒体氧化磷酸化过程中产生的有毒物质,过量的ROS 可引起细胞核DNA(Nuclear DNA,nDNA)和mtDNA 氧化损伤。 ROS 可破坏线粒体功能,导致呼吸酶活性紊乱和线粒体去极化。线粒体氧化应激增加会导致磷脂过氧化, 细胞色素氧化酶活性降低,ATP 产生减少,并导致神经元死亡[16]。 当ROS 出现病理性增多时, 超过抗氧化防御能力,导致氧化应激, 并进一步改变Ca2+稳态, 当线粒体因Ca2+超负荷时,会使线粒体通透性转变,导致外线粒体膜的渗透膨胀和破裂, 细胞内Ca2+浓度的储蓄升高可导致神经元变性和细胞死亡[16]。此外,线粒体中产生的过量ROS 也会导致mPTP 的打开,并释放细胞色素C 启动细胞凋亡。 在AD 患者大脑中观察到线粒体超氧化物歧化酶 (Superoxide Dismutase,SOD) 亚型SOD2 缺失,SOD2 的表达下调导致大脑内Aβ 水平升高[17],以及Aβ 诱导的ROS 过度产生修饰细胞信号通路, 并通过p38 丝裂原活化蛋白激酶(MAPK)启动tau 蛋白过度磷酸化,高磷酸化tau蛋白的异常积累可导致细胞内神经纤维缠结(Neurofibrillary Tangles, NFT)的形成[18],SOD1 的突变导致ROS 通过表达一氧化氮合酶导致肌萎缩性侧索硬化症(Amyotrophic Lateral Sclerosis, ALS)的病理改变[19-20]。 越来越多的证据表明,在神经退行性疾病患者大脑中可观察到高水平的ROS,并伴有线粒体功能障碍[21-22]。
1.4 线粒体质量控制缺陷
线粒体稳态对正常的神经元功能至关重要,通过监测线粒体损伤,去除受损线粒体,以及维持正常线粒体合成,可以保证线粒体的正常形成与功能,细胞对线粒体质量及有缺陷线粒体的监测过程称为线粒体质量控制[23]。 线粒体质量控制在神经退行性疾病病理过程中发挥重要的作用。 AD 患者的成纤维细胞的细胞核周围聚集大量形态细长的线粒体,同时Drp1 表达水平下降; 而AD 相关蛋白早老素(PS1)的突变可升高溶酶体PH 值,从而降低其水解酶能力,抑制自噬体的清除作用;在PTEN 诱导激酶1(PTEN-Induced Kinase1, PINK1) 敲除的哺乳动物神经元细胞中,线粒体中Ca2+浓度增加,导致ROS 产生增多,最终出现细胞死亡[24-25]。 线粒体自噬能力下降促进α- 突触核蛋白的沉积,其聚集成低聚物并导致细胞死亡; 在HD 患者的脑组织中线粒体碎片增多、氧化呼吸功能受损。 另外,体内和体外实验都表明,亨廷顿蛋白(Hungting Protein, Htt)突变会阻碍Ca2+缓冲能力,从而降低线粒体膜电位,损害氧化磷酸化过程,导致线粒体损伤[26];当受损的线粒体没有被线粒体质量控制途径修复并且无法维持功能时,会导致mPTP 孔打开,释放细胞色素C,与凋亡蛋白酶1 (Apoptotic Protease-Activating Factor-1, Apaf-1)结合形成凋亡体并介导细胞凋亡, 而过度细胞凋亡与神经退行性疾病的发病机制直接相关[27]。 综合以上研究表明, 缺陷的线粒体质量在神经退行性疾病的发病机制中至关重要。
2 神经退行性疾病与线粒体功能障碍
在有神经退行性疾病患者的大脑中, 线粒体ATP 合成和能量产生减少,ROS 增加,Ca2+缓冲能力下降,以及MPTP 开放,大脑出现线粒体功能障碍[28]。越来越多的证据表明, 线粒体功能障碍在神经退行性疾病发病机制中发挥着重要作用[29]。因此,了解线粒体在神经退行性疾病发病机制中的作用对于开发治疗措施非常重要(表1)。

表1 最常见的神经退行性疾病中线粒体功能障碍的主要原因Table1 Main causes of mitochondrial dysfunction in the most common neurodegenerative diseases
3 运动促进线粒体功能改善神经退行性疾病的分子机制
3.1 运动与神经退行性疾病
运动是与衰老相关的神经退行性疾病线粒体功能障碍的有效治疗措施,运动可减缓认知能力下降,改善突触功能,促进生存因子合成[30]。运动还通过减少细胞凋亡和神经胶质细胞活化增加神经保护作用。 运动产生的这些改变主要通过增加线粒体功能实现(图2),包括:①运动通过溶酶体降解和线粒体质量控制缓解与衰老相关的认知能力下降[33];②运动通过增强线粒体动力学和电子传递链功能促进大脑线粒体功能[34];③运动还通过参与氧化还原调节、线粒体生物能量学、生物合成、动力学、质量控制和凋亡信号传导的途径提供神经保护[35];④减轻大脑皮层和小脑线粒体损伤、 氧化应激及细胞凋亡相关信号蛋白表达改变[36];⑤运动还通过增加线粒体密度增加大脑可塑性[37]。
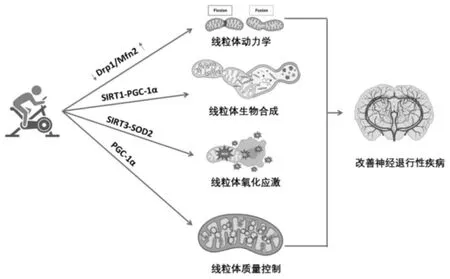
图2 运动通过调节线粒体功能障碍相关信号通路改善神经退行性疾病Figure2 Exercise improves neurodegenerative diseases by regulating mitochondrial dysfunction-related signal pathways
3.2 运动、Drp1/Mfn2 与线粒体动力学
有研究表明跑台运动与自主运动两种运动模式均上调了大脑中Mfn2 的含量, 并降低了两个大脑区域的Drp1 水平[38],但也有研究表明3 周的跑台运动导致老年小鼠皮层的Drp1 水平显著增加,但不会影响线粒体生物合成或其他应激反应途径[39]。16~18 个月的雄性大鼠进行8 周的游泳训练, 训练组与久坐不动组比较发现, 训练组大鼠海马体的Mfn2 和Drp1 水平显著高于久坐不动组,结果表明运动增加衰老海马体中的线粒体融合和裂变[31]。 Koo 等[32]报道, 长期高脂肪饮食的SD 大鼠海马线粒体融合减少、裂变增多,线粒体动力学被破坏,然而8 周的跑步机运动降低了海马中线粒体裂变蛋白Drp1 和Fis1 的表达, 同时线粒体融合蛋白Mfn1/2 和OPA1表达增加, 预防和改善了脑海马组织中线粒体功能障碍,缓解了由于肥胖引起的大脑病理变化。 Jeong等[40]研究表明8 周的跑步机练习降低了衰老SD 大鼠海马线粒体裂变蛋白Fis1、Drp1 的表达, 增加了线粒体融合蛋白OPA1、Mfn1/2 的表达,维持了大鼠海马线粒体动力学的平衡, 而单独使用线粒体靶向抗氧化剂(MitoQ)治疗的SD 大鼠改善了衰老对炎症和抗氧化酶的影响, 但对于线粒体动力学或认知功能并没有显著变化, 这也表明了运动可以通过Drp1/Mfn2 改善线粒体动力平衡保护与衰老相关的认知能力下降以及神经退行性病变。
3.3 运动、SIRT-PGC-1α 与线粒体生物合成
线粒体生物合成减少引发线粒体功能障碍,并导致衰老和神经变性, 因此增加线粒体生物合成是对神经退行性疾病过度裂变和降解的重要补偿机制。线粒体生物合成主要通过增加沉默信息调节剂1(SIRT1)的转录共激活剂来促进,SIRT1 脱乙酰化并激活PGC-1α 从而增加其转录水平[41]。 PGC-1α 在大多数组织中都有表达, 目前是公认的线粒体生物合成的有效刺激剂, 也是抗氧化和神经保护的重要信号通路。 SIRT1-PGC-1α 轴是神经退行性疾病非常重要的代偿机制,可以减少线粒体过度裂变和降解,并通过促进线粒体生物合成增加对神经的保护。Steiner 等[42]报道,8 周的耐力训练上调大脑中的PGC-1α和SIRT1 mRNA 表达,且mtDNA 表达也明显增加,这提示线粒体生物合成增加, 线粒体功能得到明显改善。 5- 腺苷单磷酸活化蛋白激酶(AMPK)是由两个调节β 亚基和γ 亚基以及一个催化α 亚基组成,是调节线粒体功能的关键因子,Bayod 等[43]研究发现,36 周的跑台运动不仅增加了SD 大鼠大脑组织PGC-1α 和SIRT1 基因表达,也激活了AMPK,并减少p53 乙酰化水平, 增加大脑中线粒体呼吸复合物含量, 这提示AMPK 可能在SIRT1-PGC-1α 调节脑线粒体生物合成中发挥着重要作用。 这些研究证据表明,运动通过激活SIRT1-PGC-1α 信号通路增加线粒体生物合成, 改善神经退行性疾病的线粒体功能障碍及损伤。
3.4 运动、SIRT3-SOD2 与线粒体ROS 和氧化应激
SIRT3 是线粒体中的主要去乙酰化酶, 在调节线粒体信号分子级联反应中起着重要作用, 它通过改变线粒体蛋白质的乙酰化状态来维持细胞代谢稳态[44]。 神经元缺乏SIRT3 常出现线粒体ROS 水平升高、Ca2+在线粒体和细胞基质中聚集。 而增强SIRT3活性可以通过调节抗氧化酶来降低ROS 水平,发挥防止氧化损伤的防御作用。MnSOD 是主要的线粒体抗氧化酶,SIRT3 直接把线粒体中SOD2 去乙酰化,显著增强了清除ROS 的能力[45]。 皮层和海马体中的MnSOD 随着年龄的增长而下降,但是Jolitha 等[46]报道成年大鼠在经过40 d 的游泳运动后,大脑区域的MnSOD 活性显著上升,氧化磷酸化水平上升,蛋白质氧化损伤被逆转。 大量的研究表明运动增加大鼠和小鼠横纹肌和大脑中SIRT3 的表达。 Cheng 等[47]报道, 小鼠通过跑轮运动增加神经元中的SIRT3 的表达, 并激活SOD2, 通过防止过度线粒体氧化应激、维持能量平衡、稳定线粒体膜结构,以及改善神经元Ca2+水平来维持线粒体功能。 Aleixo 等[38]研究表明模拟强迫耐力训练和自愿体育活动这两种耐力训练均能上调SIRT3 和MnSOD 水平, 通过减少ROS 的产生上调抗氧化防御系统来改善氧化还原平衡, 从而达到保证神经线粒体磷酸化系统的完整性和功能, 并且氧化应激增加后也会导致mPTP 开放,从而导致神经细胞凋亡,两种运动均能增强大脑和小脑中对mPTP 开放的抵抗力, 降低了凋亡标志物的表达(半胱天冬酶、Bax/Bcl2 比率),这就表明长期运动可以改善线粒体功能障碍, 对预防和改善神经系统疾病具有重要作用。
3.5 运动、PGC-1α 与线粒体质量控制
大量研究表明, 神经退行性疾病的发病机制涉及PGC-1α 介导的线粒体质量控制,PGC-1α 是Nrf1和Nrf2 的共激活因子[48]。 在一些主要的神经退行性疾病中,如HD、AD、PD,PGC-1α 的表达水平和活性被下调, 因此通过增加PGC-1α 表达来调节线粒体质量控制, 从而减少神经损伤。 在分子水平上,PGC-1α 通过调节线粒体未折叠蛋白反应(Mitochondrial Unfolded Protein Response,mtUPR)来增强蛋白酶活性,从而重新折叠或降解错误折叠的蛋白质,这可以防止由受损蛋白质的大量积累引起的线粒体功能障碍,并减少神经元损伤[49];在器官水平上,PGC-1α调节Drp1 和Mfn2 的表达, 减少海马体区域的过度线粒体裂变,促进线粒体融合[50],从而在调节神经元线粒体裂变和融合之间的平衡中发挥作用; 在细胞器水平上,由PGC-1α-PPARγ-Nrf2-NDP52 途径激活的线粒体自噬有助于降低AD 的发病机制[51]。
4 小结
运动是预防和缓解神经退行性疾病的有前途的治疗策略之一,运动通过调节线粒体生物合成、线粒体ROS 和氧化应激、线粒体融合和裂变、线粒体质量控制这几种机制, 增加大脑对衰老或神经变性的抵抗力。同时,线粒体功能障碍也是衰老和神经变性的重要病理改变, 所以了解由运动诱导的大脑线粒体功能改善机制将提供新的和更有效的神经退行性疾病治疗策略。 但由于运动方式和运动强度的多样性和复杂性,不同运动方式、强度和持续时间对大脑线粒体的刺激及调控机制有待进一步探索。另外,在不同年龄段、不同类型的神经退行性疾病患者的运动康复治疗中, 如何选择最佳的运动方式来保证运动对大脑线粒体不同机制的调节,需要更深入的研究。
