Kaemtakols A-D,highly oxidized pimarane diterpenoids with potent anti-inflammatory activity from Kaempferia takensis
2023-12-29OrawanJongsomjainukJutatipBoonsombatSanitThongnestHunsaPrawatParatchataBatsomboonSitthivutCharoensutthivarakulSarojRuchisansakunKittipongChainokJitnapaSirirakChulabhornMahidolandSomsakRuchirawat
Orawan Jongsomjainuk ,Jutatip Boonsombat,4 ,Sanit Thongnest,4* ,Hunsa Prawat,4 ,Paratchata Batsomboon ,Sitthivut Charoensutthivarakul ,Saroj Ruchisansakun,Kittipong Chainok,Jitnapa Sirirak,Chulabhorn Mahidol,3 and Somsak Ruchirawat,3,4
Abstract Four highly oxidized pimarane diterpenoids were isolated from Kaempferia takensis rhizomes.Kaemtakols A-C possess a tetracyclic ring with either a fused tetrahydropyran or tetrahydrofuran motif.Kaemtakol D has an unusual rearranged A/B ring spiro-bridged pimarane framework with a C-10 spirocyclic junction and an adjacent 1-methyltricyclo[3.2.1.02,7]octene ring.Structural characterization was achieved using spectroscopic analysis,DP4+and ECD calculations,as well as X-ray crystallography,and their putative biosynthetic pathways have been proposed.Kaemtakol B showed significant potency in inhibiting nitric oxide production with an IC50 value of 0.69 μM.Molecular docking provided some perspectives on the action of kaemtakol B on iNOS protein.
Keywords Kaempferia takensis,Diterpenoid,Structure elucidation,Anti-inflammatory,DP4+,Molecular docking
1 Introduction
The genusKaempferiais a member of the Zingiberaceae family,encompassing over 60 species.This genus exhibits a broad distribution across Southeast Asia,with a significant presence in Thailand [1,2].The rhizomes ofKaempferiaplants are rich in isopimarane,abietane,labdane,and clerodane type diterpenoids[3-6].Additionally,some intriguing rearranged diterpenoids have been identified,such as unique norditerpenoids featuring an epoxide bridge with a hemiketal ring named elegansins D and E,fromK.elegans[7].
Compounds isolated fromKaempferiaspecies have gained attention due to their wide array of structural variations and remarkable anti-inflammatory effects[8-11].Nonetheless,the scientific exploration concerning chemical characterization and biological studies has been confined to only a handful of species,such asK.parvifera,K.galanga,K.marginata,andK.pulchra[1].As a result,research on plants in this genus,essentially those unexplored,could potentially unveil novel diterpenoids with intriguing structural frameworks and promising biological properties.
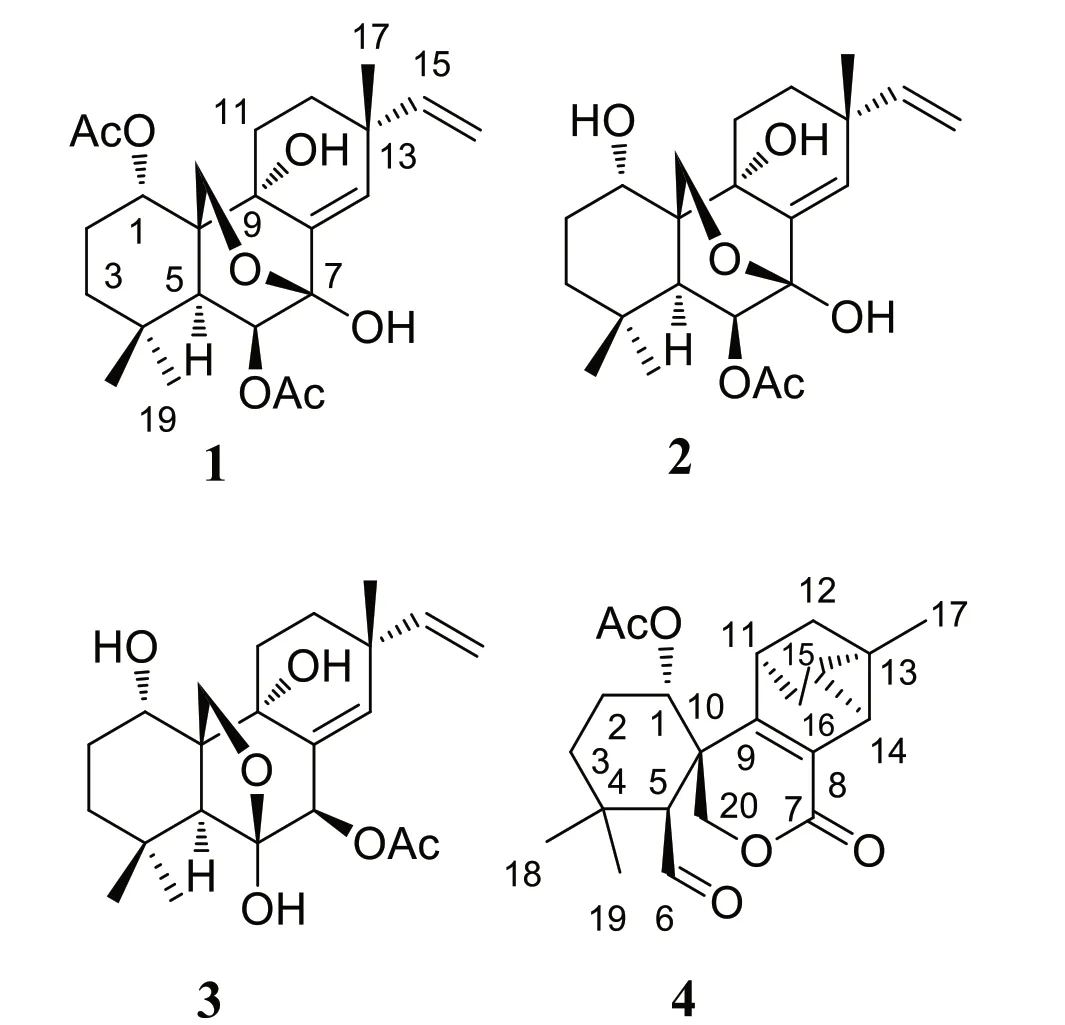
Fig. 1 Structures of compound 1-4
Here,we investigated the chemical constituents fromK.takensisBoonma & Saensouk,which to date have not yet been reported.Our investigation led to the isolation of the architecturally unique highly oxidized pimarane diterpenoids,kaemtakols A-D (1-4,Fig.1).Compounds1-3were characterized as C-20 oxygenated methylene pimarane-type diterpenoids with either a fused tetrahydropyran or tetrahydrofuran ring.Compound4featured an unusual rearranged A/B ring spiro-bridged pimarane framework with the presence of a rare spiro[5,5]decane moiety adjacent to a 1-methyltricyclo[3.2.1.02,7]octene ring.All compounds were examined for their ability to reduce inflammation,by suppressing nitric oxide (NO) generation in RAW264.7 macrophage cells and by inhibiting NF-κB production in HaCat human skin cells.
2 Results and discussion
Kaemtakol A (1),colorless crystals,has a molecular formula C24H34O7,deduced from the HRESIMS [M+Na]+atm/z457.2204 and supported by13C and DEPT 135 NMR data,suggesting eight unsaturation indices.The IR absorption peaks at 3415 cm-1and 1740 cm-1indicated hydroxy and ester functional groups.The1H-NMR(Table 1) exhibited signals for three quaternary methylsδH/δC1.09/25.4 (CH3-17),1.37/22.6 (CH3-18),and 1.08/34.5 (CH3-19);two acetyl methyls atδH2.12 and 2.22;an oxymethylene group atδH/δC3.53 and 3.96 (each 1H,d,J=10.4 Hz)/65.8;two oxymethine groups atδH/δC4.78/71.9 and 5.64/71.7;a vinyl group with theδHvalues of 4.98 (1H,d,J=10.6 Hz),5.04 (1H,d,J=17.6 Hz),and 5.86 (1H,dd,J=17.5,10.6 Hz);and an olefinic proton atδH/δC5.95/132.9.The13C-NMR and DEPT of compound1presented twenty-four carbon signals,which matched with five methyl carbons including two acetyl methyls,six methylene carbons including one alkene and oneoxygenated,five methine carbons including two oxymethines and two olefinics,and eight quaternary carbons including one alkene,two carbonyls,one oxygenated,and one hemiketal (Table 1).Excluding the above four degrees of unsaturation from two double bonds and two carbonyls,the remaining four unsaturation indices implied a tetracyclic compound.Hence,the structure of1was identified as C-20 oxygenated methylene pimarane diterpenoid,featuring a tetracyclic ring bearing two acetoxy groups.The COSY correlations of four spin systems comprising H-1/H2-2/H2-3,H-5/H-6,H2-11/H2-12,and H-15/H2-16,together with the HMBC correlations connecting H3-18 and H3-19 to C-3,C-4,and C-5; H2-20 to C-1,C-5,C-7,and C-9;H-1 to C-3,C-5,and C-20;H-5 to C-6,C-10,C-18,C-19,and C-20;H-14 to C-7,C-9,C-13,C-15,and C-17;and H3-17 to C-12 and C-14,suggested the structural architecture of1(Fig.2).Based on the NOESY experiment,it was indicated that rings A and B displayed atrans-decalin,with theα-axial hydrogen on C-5 and aβ-axial position of C-20.Additionally,the acetoxy group on C-1 was found to be in anα-axial position,and the oxygen linkage connecting C-7 and C-20 was situated above the B-ring plane.The C-6 relative configuration was assigned by a large coupling constant value of3J5,6(≈ 10 Hz) which implied that the dihedral angle between them was nearly 0 degree,indicating acisrelationship [12].This relationship was further proved by the presence of H-5 and H-6 with H3-19 correlations,and the absence of correlation of either H3-18 or H2-20 in the NOESY spectrum (Fig.2).Thus,these above observations revealed theβ-equatorial position of the OAc group on C-6.The X-ray crystallography analysis supported the assigned conformation of1and the visual representation of the crystal structure provided by ORTEP drawing is depicted (Fig.3).Consequently,1was structurally determined as 7α,9α-dihydroxy-1α,6β-diacetoxy-7β,20-epoxyisopimara-8(14),15-diene.Furthermore,based on the high resemblance between the calculated and measured ECD spectrum,the (1S,5S,6S,7R,9S,10S,13R) absolute configuration of1was determined (Fig.4).
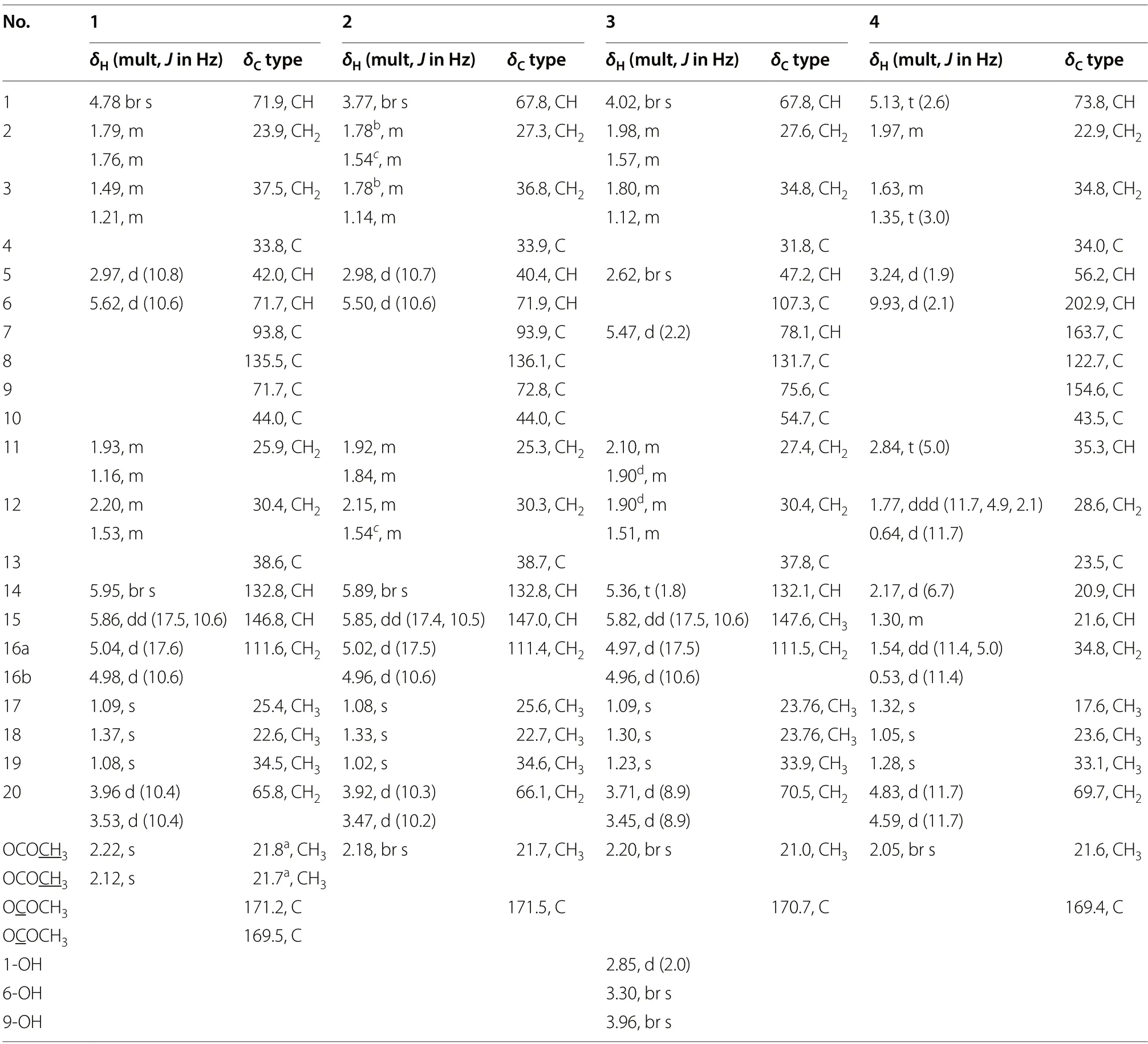
Table 1 1H (400 MHz) and 13C (100 MHz) NMR spectroscopic data of 1-4 in CDCl3
A white amorphous compound2named kaemtakol B was formulated as C22H32O6by HRESIMS atm/zof 415.2099 [M+Na]+.NMR analysis revealed that compounds1and2were C-20 oxygenated methylene pimarane-type diterpenoids bearing an epoxide ring (Table 1).Their NMR data closely resembled each other,with the only difference being the presence of a secondary alcohol on C-1 in2.This alcohol at C-1 (δC67.8) position was confirmed using HMBC cross-peaks that connected H-1 (δH3.77) to C-5,and H2-20 to C-1 and C-5.Additionally,an oxygen linkage connecting C-7 and C-20 resembling that of1could be established from HMBC cross-peaks connecting H-1 and H-5 to C-20;and H-5,H-14,and H2-20 to C-7.The above analyses enabled the establishment of the 2D structure of2.The NOESY experiment (Fig.2) unveiled an identicaltrans-decalin A/B ring pattern to1,with the hydroxy group on C-1 occupying anα-equatorial position,the 7,20-oxygen bridge situated above the B-ring plane,HO-7 positioned in anα-orientation,and H2-20 in part of the 7,20-oxygen bridge located in aβ-orientation.Consequently,2was structurally determined as 1α,7α,9α-trihydroxy-6βacetoxy-7β,20-epoxyisopimara-8(14),15-diene.The high resemblance between the calculated and measured ECD spectra of2strongly suggested the (1S,5S,6S,7R,9S,10S,13R) absolute configuration (Fig.4).
The 1D-NMR spectra of kaemtakol D (3) suggested the presence of a C-20 oxygenated methylene pimarane diterpenoid,and its structure appeared to resemble that of2(Table 1).The main differences were detected in the placement of the C-6 hemiketal hydroxy group atδC107.3 and the C-7 acetoxy group atδC78.1.These differences were confirmed through HMBC cross-peaks connecting H-5 and H2-20 to C-6,and H-5 and H-14 to C-7(Fig.2).NOESY interactions and the optimized structure obtained from the conformer analysis were used to determine compound3relative configuration (Fig.2),which suggested thetrans-decalin arrangement of the A/B rings having H-5 positioned in anα-axial orientation and C-20 in anβ-axial orientation.As a consequence,theβ-oriented acetoxy group at C-7,theα-oriented hydroxy group at C-1,and the oxygen linkage connecting C-6 and C-20 above the B-ring plane were assigned.Hence,compound3was determined to be 1α,6α,9α-trihydroxy-7βacetoxy-6β,20-epoxyisopimara-8(14),15-diene.The high resemblance between the calculated and measured ECD spectra of3established the (1S,5S,6S,7R,9R,10S,13R)absolute configuration (Fig.4).
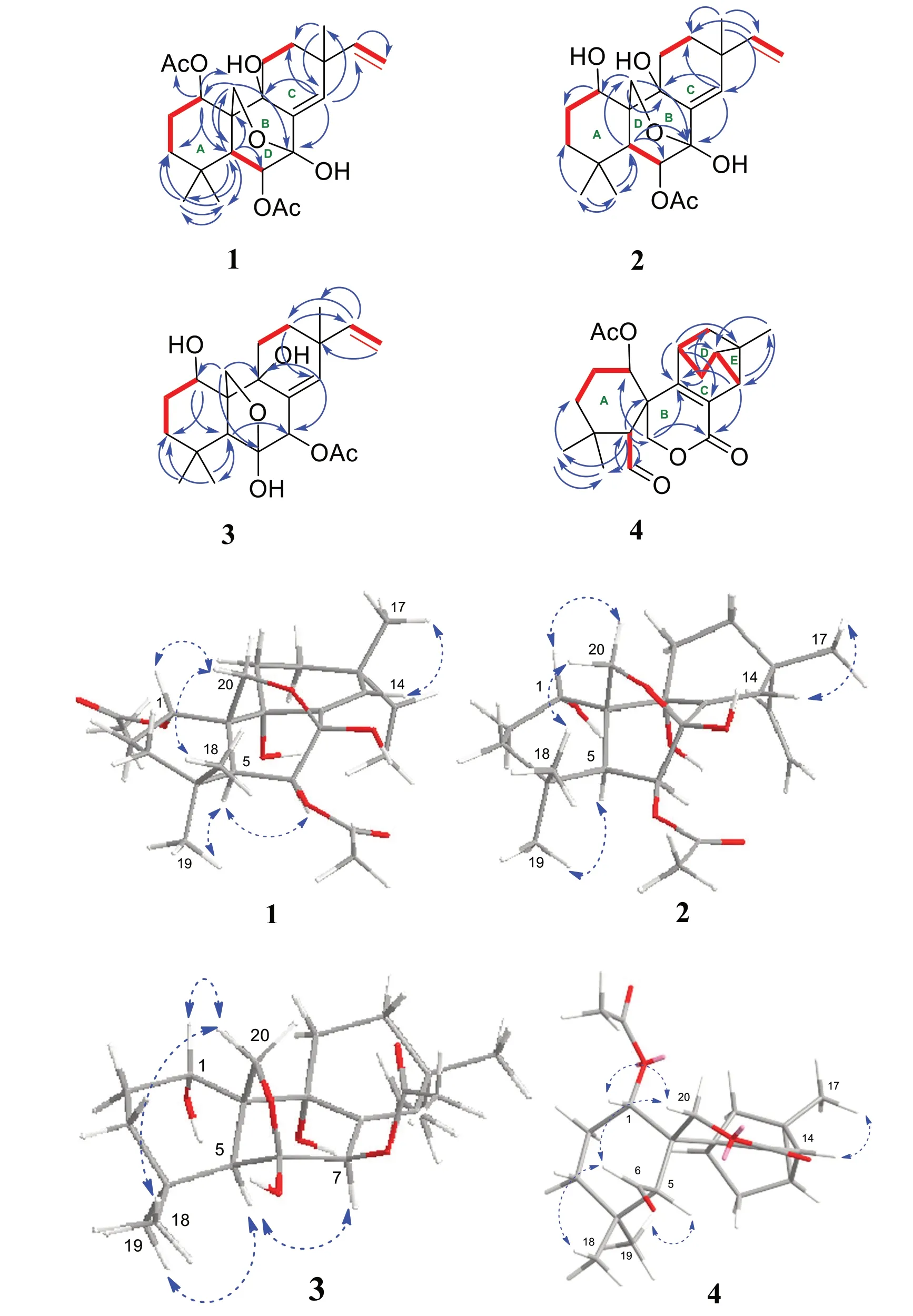
Fig. 2 Key COSY (red dash),HMBC (blue curved arrow),and NOESY (blue dotted curve arrow) correlations of compounds 1-4
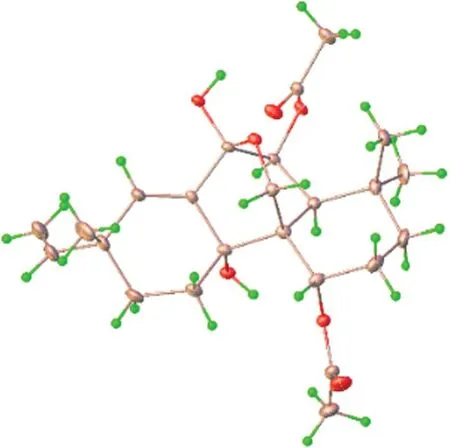
Fig. 3 Crystal structure of compound 1
Kaemtakol D (4) presented a molecular formula C22H28O5,deduced from the HRESIMS ion atm/z395.1827 [M+Na]+,consistent with nine unsaturation indices.The IR absorption peaks at 3352,1732,and 1714 cm-1suggested hydroxy and carbonyl functional groups.The13C-NMR data revealed twenty-two carbon signals as the resonances of four singlet methyls (one of which being an acetoxy),five methylenes (one of which being an oxymethylene carbon),six methines (including one oxymethine carbon and one sp2hybridized aldehyde carbon),and seven quaternary carbons (including one olefinic carbon and two sp2hybridized carbonyls).The abovementioned functional groups attributed to four out of the nine degrees of unsaturation,leaving five degrees to be deduced as a pentacyclic ring.The1H-NMR spectrum exhibited four methyl protons atδH2.05 (an acetyl group),1.32,1.28,and 1.05,oxymethylene protons atδH4.83 and 4.59,an oxymethine proton atδH5.13,as well as an aldehyde proton atδH9.93 (Table 1).The COSY spin systems led to the assignment of three fragments: H-1/H2-2/H2-3 for C-1 to C-3,H-5/CHO for C-5 with an aldehyde function,and H-14/H-15/H2-16/H-11/H2-12 for C-11 to C-16 (Fig.2).By deduction from HMBC correlations (Fig.2),these fragments expanded to form the major portion of the molecules.Furthermore,the HMBC correlations were used to establish the bond connections between rings A and B,whereby the observed crosspeaks from H-5 to C-1 (δC73.8),C-6 (δC202.9,an aldehyde group),C-10,C-20 (δC69.7),and a gem-dimethyl group,from H-6 to C-5,from a gem-dimethyl group to C-3 and C-5,and from H2-20 to C-1,C-5,C-7 (δC163.7,an ester carbonyl group),C-9 (δC154.6,an alkene group),and C-10,indicated the appearance of a six-membered carbocycle (A) with a geminal dimethyl group at C-4,an aldehyde unit at C-5,a lactone ring (B),two oxygen atoms(at C-1 and C-20),along with two quaternary carbons (at C-4,and C-10).Therefore,rings A and B are united by a bridgedspirolactone moiety.The incorporation of the remaining tricyclo unit to complete a pentacyclic structure was guided by the HMBC cross-peaks connecting H-11 to C-8 and C-13,H2-12 to C-9,and H-14 to C-9 and C-17 (Fig.2).In addition,an isolated methylene unit that displayed an AB geminal coupling pattern of H2-16 (δH1.54 and 0.53) was assigned to be at the junction between C-11 and C-13,and this position was further verified by long-range correlations between H-11 and C-15,H2-16 and C-9,H2-16 and C-12,as well as H3-17 and C-14 and C-15.Moreover,the cross-peaks from H-14 to C-7(δC163.7) signified the incorporation of the carbonyl carbon into the lactone ring,thereby necessitating the linkage of C-7 to C-8.The last remaining assignment of carbonyl carbon for the acetate group (OAc,δC169.4)was positioned at C-1 due to a higher chemical shift value (δC73.8) in the downfield region,compared with C-1 in compounds2and3(67.8 ppm).The compound4relative stereochemistry was assigned using the NOESY correlations of H3-18/H-6,H3-19/H-5,and H-5/H-3α(Fig.2).Further evidence for theα-configuration of the C-1 acetoxy group was provided by NOESY cross-peaks among H-1,H-6β,and H2-20,which inferred their cofacial relationship.The location of C-13 was deduced from strong correlations connecting H-12βand H-14 to H3-17β,and the lack of correlation between H-11 and either H-14 or H-15 in the NOESY spectrum.Based on these considerations,4was characterized as 1α-acetoxy-13β-methyl-6,7-seco-7,20-olide-8,9-tricyclo[3.2.1.02,7]octane-isopimara-8-en-6-al.
The relative configuration of the carbon positions 5,10,11,13,and 14 in4was determined by DP4+NMR chemical shift probability analysis.1H and13C chemical shifts for four possible isomers (4and4a-4c) were computed and compared to the experimental values(see Additional file 1).The statistical analysis confirmed the agreement with isomer4,with a 100% probability.Furthermore,the high resemblance between the calculated and measured ECD spectra of4established the(1S,5S,10S,11R,13R,14S,15S) absolute configuration(Fig.4).
It is noteworthy that compound4is partially associated with trachylobane diterpenoids [13,14],known for their characteristic cyclopropane moiety.However,4stands out from this structural family by incorporating two additional functional elements,which are the adjacent tricyclo[3.2.1.02,7]octene ring system [15,16] and a spirocyclic lactone ring junction at C-10,thereby expanding the diversity of this particular structural type.
The putative biosynthetic pathway of compounds1-4is illustrated in Fig.5.Compounds1-4are possibly formed from geranylgeranyl pyrophosphate (GGPP)through the pimara-8(14),15-diene (I).Oxidation at multiple sites ofIgives intermediateII,which is presumably a common biosynthetic precursor of compounds1-4.Further oxidation at C-6 ofIIfollowed by ketal formation givesIV,which is then acetylated at 7-OH to give3.Alternatively,oxidation at C-7 ofIIfollowed by ketal formation provides intermediateVI.Acetylation at different sites ofVIyields compounds1and2and intermediateVII.Compound4is proposed to originate fromVII,whereby the glycol cleavage followed by dehydration generates the diene moiety inIX.Upon intramolecular Diels-Alder reaction with the adjacent double bond inIX,compound4is formed.

Fig. 4 Simulated and calculated ECD spectra of 1-4
Kaemtakols A-D (1-4) were screened for their inhibitory activity in suppressing the NO production triggered by lipopolysaccharide in RAW 264.7 macrophage cells and the NF-κB production activated by cytokine-induced inflammation in HaCat human skin cells.The results(Table 2) showed that2potently inhibited NO production having an IC50of 0.69 μM.In contrast,substituting the acetylated group at position 1 of compound2,namely kaemtakol A (1),abolished anti-inflammation activity(IC50=103.9 μM).In addition,compounds1-3satisfyingly exhibited minimal cytotoxicity towards RAW 264.7 cells at the specific concentrations needed to inhibit NO production.Nevertheless,these compounds did not show discernible inhibitory effects against NF-κB(EC50> 100 μM).
To determine the possible binding mode of compound2to iNOS,docking studies were conducted using the crystal structure with high-resolution iNOS protein(PDB 3E6T).Results (Fig.6a,b) revealed that the binding position of2was next to cofactors: HEM-901 and H4B-902,and overlapped partly with that of the iNOS inhibitor AR-C118901 [17].Compound2exhibited a binding energy of -8.0 kcal/mol,which was marginally greater than that of AR-C118901 (-9.2 kcal/mol),indicating that2and the cocrystal inhibitor had roughly equivalent binding affinities (Table S16).Moreover,2occupied the binding pocket through hydrophobic interactions and hydrogen bonds with crucial amino acid residues,including GLN257,ARG260,TYR341 and ARG260 (Fig.6c).As depicted in Fig.6d,the hydroxy and acetoxy groups of2played an important role in hydrogen contacts with GLN257 and ARG260,while its alkene and methyl groups took part in the hydrophobic interactions with MET114,TRP84,TYR341,and TYR367 in the cavity of iNOS.

Fig. 5 Putative Biosynthetic Pathways to Generate 1-4

Table 2 Inhibitory effects of the compounds 1-4 against the LPS-induced NO production in RAW264.7 cells and NF-κB production by cytokine induced inflammation in HaCat human skin cells
3 Experimental procedures
3.1 General experimental procedures

Fig. 6 The binding position of compound 2 (pink) in a the whole receptor containing AR-C118901 (green) and cofactor: HEM-901 and H4B-902(blue),and b the binding pocket of iNOS.c Potential hydrogen contacts (green dash) and hydrophobic interactions (pink dash) of compound 2 with amino acid residues in the binding site of iNOS.d 2D diagram representing interactions of compound 2 in the cavity of iNOS (PDB ID: 3E6T)
Column gel filtration involved using Sephadex LH-20(GE Healthcare) and MCI gel (75-150 μm;Mitsubishi Chemical,Tokyo),as well as RP-C18and silica gel 60 from Merck for column chromatography separation.HPLC analysis was carried out using a Waters Delta 600 with a Waters 2996 photodiode array detector.NMR spectra were collected using Bruker Avance 400 and 600 spectrometers.HRESIMS analysis was performed with a Bruker Daltonics microTOF spectrometer.A PerkinElmer Spectrum One Spectrometer using a universal attenuated reflectance (ATR) technique was utilized to record the IR spectra.A JASCO P-1020 polarimeter was employed for recording optical rotation,and A JASCO J-810 spectropolarimeter for recording CD spectra.
3.2 Plant materials
In April 2022,the whole plants ofKaempferia takensisBoonma & Saensouk were collected in Tak Province,Thailand.Assist.Prof.Dr.Saroj Ruchisansakun provided the identification.The Suan Luang Rama IX Botanic Garden in Thailand housed a reference specimen (S.R.1851).
3.3 Extraction and isolation
The freshK.takensisrhizomes (414.6 g) were soaked in two rounds with an equal volume (1.5 L) of 95% EtOH and 50% dichloromethane-MeOH mixture at ambient temperature,respectively.The complete evaporation of the solvent gave respective crude extracts of 7.04 g and 1.53 g.Due to these two crude extracts exhibiting the same NMR patterns of diterpenoids,we decided to combine them to yield 8.6 g of a crude extract in order to get more pure active compounds.
The combined crude extract (8.6 g) was processed to the Sephadex LH-20 column using a dichloromethane-MeOH (1:1) elution,resulting in four fractions (F1-F4).F3 (2.8 g) was subjected to further separation on a Sephadex LH-20 column,and elution was carried out using a dichloromethane-MeOH (60:40) mixture to obtain four subfractions (Fr3.1-Fr3.4).Fr.3.2 (289.4 mg) was purified using semi-preparative HPLC with a Hichrom C18(250 × 21 mm i.d.).Elution was carried out with a gradient of 50:50-95:5 MeCN in H2O for 90 min at a flow rate of 10 mL/min,monitored at UV wavelength λ 220 nm.This process yielded 1.4 mg of kaemtakol D (4,tR25 min).Fr.3.3 (287.0 mg) underwent purification using preparative HPLC with a YMC-Pack ODS-A C18 column(250 × 20 mm i.d.).The purification was achieved through gradient elution with 50-95% MeCN in H2O for 45 min at a flow rate of 10 mL/min,monitored at a UV wavelength of λ 220 nm.This process yielded 28.0 mg of kaemtakol A (1,tR16 min).Fr.3.4 (2.3 g) was subjected to gel filtration with Sephadex LH20,using 100% MeOH as the eluent,resulting in the separation into six subfractions(Fr.3.4.1-3.4.6).Subsequently,Fr.3.4.2 (2.0 g) underwent a second round of separation by Sephadex LH20,again using 100% MeOH as the eluent,yielding six additional subfractions (Fr.3.4.2.1-3.4.2.6).Fr.3.4.2.3 (1.7 g)was additionally purified by semi-preparative HPLC(Hichrom C18250 × 21 mm i.d.) with 50-95% MeCN in H2O gradient elution over 90 min at a flow rate of 10 mL/min,monitored at UV λ 220 nm.This process yielded kaemtakol B (2,140.0 mg,tR12 min) and kaemtakol C (3,43.0 mg,tR14 min).
3.3.1 Kaemtakol A (1)
Colorless crystals; C22H32O6,mp 205-208 °C;+40.6(c0.59,MeOH);UV (MeOH)λmax(logε): 279 (0.838) and 212 (2.916) nm;CD (MeOH)λmax(Δε) 222 (-87.9289)nm;IR (ATR)νmax3415,2953,2869,1740,1608,1372,1227,1026,910,829,and 733 cm-1;1H (400 MHz) NMR data (CDCl3) and13C (100 MHz) NMR data (CDCl3) see Table 1;ESI HRMSm/z457.2204 [M+Na]+(calcd for C24H34O7Na,457.2197).
3.3.2 Kaemtakol B (2)
White amorphous; C22H32O6,+43.5 (c0.15,MeOH);UV (MeOH)λmax(logε) 245 (0.36) nm;CD (MeOH)λmax(Δε) 343 (-11.6695),255 (-14.299),220 (-63.4363) nm;IR (ATR)νmax3413,2935,1734,1365,1235,1023,911,829,and 729 cm-1;1H (400 MHz) NMR data (CDCl3)and13C (100 MHz) NMR data (CDCl3) see Table 1;ESI HRMSm/z415.2099 [M+Na]+(calcd for C22H32O6Na,415.2091).
3.3.3 Kaemtakol C (3)
White amorphous; C22H32O6,+43.1 (c0.38,MeOH);UV (MeOH)λmax(logε) 214 (0.30) nm;CD(MeOH)λmax(Δε) 225 (-10.8958) nm,215 (-15.1665)nm,196 (+3.7076) nm;IR (ATR)νmax3375,2943,1725,1636,1371,1237,1191,1047,960,891,and 735 cm-1;1H (400 MHz) NMR data (CDCl3) and13C (100 MHz)NMR data (CDCl3) see Table 1;ESI HRMSm/z415.2097[M+Na]+(calcd for C22H32O6Na,415.2091).
3.3.4 Kaemtakol D (4)
White amorphous; C22H28O5,+182.1 (c0.08,MeOH);CD (MeOH)λmax(Δε) 264 (+13.7087) nm,233 (+6.6737) nm;IR (ATR)νmax3352,2927,2866,1732,1714,1655,1374,1236,1115,1063,and 764 cm-1;1H (400 MHz) NMR data (CDCl3) and13C (100 MHz)NMR data (CDCl3) see Table 1;ESI HRMSm/z395.1827[M+Na]+(calcd for C22H28O5Na,395.1829).
3.4 X-ray crystallographic analysis of 1
Colorless crystals of kaemtakol A (1) were obtained using the vapor diffusion method from a solution of MeOHEtOH (1:1) and MeOH.X-ray crystallographic analyses were conducted at 150 K using a Bruker APEX-II CCD diffractometer with Mo Kα radiation.The crystallographic data was submitted to the Cambridge Crystallographic Data Center (CCDC number 2247367).The data is accessible at no cost through www.ccdc.cam.ac.uk/data_ reque st/ cif,or by emailing data_request@ccdc.cam.ac.uk,or by contacting The Cambridge Crystallographic Data Centre,12 Union Road,Cambridge CB2 IEZ,UK,fax:+44 1223 336,033.
3.5 ECD calculations and DP4+analysis
The computation methods employed in this study were slightly modified from previous reports [18].A conformational search within a 5 kcal/mol energy window was conducted using Spartan’ 20 with the MMFF94 molecular mechanics model.The Gaussian 16 Rev.C.01 package was utilized for DFT calculations [19,20].All low-energy conformers were additionally optimized at the ωB97XD/cc-PVDZ level of theory with the IEFPCM solvent model(methanol).At the same computational level,the vibrational frequencies of all optimized conformers were analyzed to verify the presence of actual electronic potential energy minima,and no imaginary frequency was detected.The low-Gibbs free energy conformer with over 2% population in the Boltzmann distribution was subjected to ECD calculations.TD-DFT calculations were performed at the M06-2x/def2-SVP level with the IEFPCM solvent model (methanol) [21-23].For each conformer,calculations were performed for 30 excited states,and the simulated ECD curves were post-processed using SpecDis.This involved applying a Boltzmann averaging approach across all conformers and employing overlapping Gaussian functions with an exponential half-width(σ=0.35) [24,25].The theoretical ECD spectra of1-4of their related diastereomers and enantiomers were directly compared with experimental ECD spectra.In addition,for the same set of low-energy conformers,magnetic shielding tensors (σ) were computed using the GIAO method at the mPW1PW91/6-31+G(d,p) level in PCM solvent modeling,and then weight-averaged.Finally,DP4+analysis was carried out using the provided Excel tool [26].CYLview was utilized for virtualizing all 3D structures [27].
3.6 Molecular docking with human iNOS
Molecular docking was performed using Autodock Vina[28,29] and AutoDockTool 1.5.6 to examine the binding of compound2in the cavity of iNOS oxygenase.The protein structure involved in the inhibition of nitric oxide synthase (NOS),iNOS oxygenase (PDB 3E6T),was acquired from Protein Data Bank (http:// www.rcsb.org/).This structure served as the receptor.The molecular structure of2was obtained through geometry optimization using Gaussian09 at the B3LYP/6-311G* level.The central grid parameters were set at 70-70-70 along the x-,y-and z-axes,with a 1.000 Å spacing to encompass all protein structures.The analysis and visualization of the docking results were performed using the BIOVIA Discovery Studio Visualizer [30].
3.7 Inhibition of NO production in LPS-activated murine macrophages (RAW 264.7 cells)
The inhibitory assay followed the methods described previously with some modifications [31,32].RAW 264.7 cells were seeded at 3 × 104cells/well in a 96-well flat bottom plate and cultured in DMEM high glucose supplemented with 10% FBS,1% penicillin-streptomycin,and 1% HEPES.The plates were initially incubated at 37 °C for 24 h under 5% CO2atmosphere.Thereafter,LPS (0.25 μg/mL) in the absence or presence of different concentrations of kaemtakols (100,10,1,0.1,0.01,and 0.001 μg/mL) were introduced into the culture plate and then incubated further for 24 h under the same condition.Aminoguanidine hydrochloride (iNOS inhibitor)was also added as a positive control.The nitrite accumulation in the media was determined by the Griess reaction.The 75 μL of each culture supernatant was mixed with an equivalent amount of Griess reagent and the resulting mixture was incubated at room temperature for additional 10 min.Then,the absorbance at 546 nm was measured using a multimode microplate reader (ENVISION).The cytotoxicity of kaemtakols against RAW 264.7 was determined by a standard MTT assay.
3.8 Assessment of inhibitory effects on NF-κB nuclear translocation in human skin HaCaT cells
The assay followed the methods described previously with some modifications [33,34].Human skin cells(HaCaT cells) were seeded at 5 × 104cells/well in a 96-well black flat-bottom plate,using DMEM (Dulbecco’s Modified Eagle Medium) high glucose supplemented with 10% FBS and 1% penicillin-streptomycin.The plates were then incubated at 37 °C for 24 h under 5% CO2atmosphere.Following the initial incubation,a range of concentrations (100,50,25,12.5,6.25,3.125 μM) of kaemtakols were introduced to the cells,and a subsequent 1-h incubation at 37 °C with 5% CO2was carried out.
Afterward,a cytokine cocktail comprising TNF-α (10 ng/mL),IL-1β (10 ng/mL),and IL-6 (10 ng/mL),along with the same concentration range of kaemtakols,was introduced to the culture plates and incubated at 37 °C with 5% CO2for additional 30 min.Thereafter,the treated cells were fixed with 100% methanol at -20 °C for 10 min.The fixed cells underwent triple washes with DPBS before being exposed to a blocking solution (3% BSA) at room temperature for 60 min.Subsequently,a primary antibody (anti-NF-κB),a Rabbit monoclonal antibody,was introduced into the culture plate at a 1:500 ratio in DPBS and incubated at 4 °C overnight.Afterward,the culture plate was washed with DPBS three times.A secondary antibody (Alexa Fluor 488-conjugated goat antirabbit IgG) at a 1:500 ratio in DPBS was added to the culture plate and incubated for 1 h at room temperature in the dark.Simultaneously,Hoechst (at a 1:1,000 ratio in DPBS) was also added to the culture plate.The translocation of NF-κB in cells treated with the cytokine cocktail and kaemtakols was subsequently detected using a highcontent imaging system (Operetta).The cell viability was determined by Hoechst DNA staining (Operetta).
4 Conclusions
In summary,unprecedented highly oxidized pimarane diterpenoids named kaemtakols A-D (1-4) were isolated fromK.takensisfor the first time.This discovery adds to the diversity of structural types of natural diterpenoids.Kaemtakol B displayed the highest potency in NO production inhibition,signifying its potential as a lead compound for novel anti-inflammatory agent development.
Supplementary Information
The online version contains supplementary material available at https:// doi.org/ 10.1007/ s13659-023-00420-0.
Additional file 1.X-ray crystal data analyses and structure refinement for1.Spectra of compounds1-4,including 1D-and 2D-NMR,ESI HRMS,CD,ECD,and IR techniques.ECD calculations and DP4+analysis.X-ray crystallographic data for compound1.Table S1DP4+probability Excel sheets of compound4.Table S2Conformational analysis of1.Table S3Conformational analysis of2.Table S4Conformational analysis of3.Table S5Conformational analysis of4.Table S6Conformational analysis of4a.Table S7Conformational analysis of4b.Table S8Conformational analysis of4c.Table S9Coordinates of Compound1.Table S10Coordinates of Compound2.Table S11Coordinates of Compound3.Table S12Coordinates of Compound4.Table S13Coordinates of Compound4a.Table S14Coordinates of Compound4b.Table S15Coordinates of Compound4c.Table S16Summary of binding energies,amino acid residue involved in the hydrogen bond and hydrophobic interactions of2observed in molecular docking studies..Figure S1ORTEP drawing of crystal structure of1.Figure S21H NMR spectrum (400 MHz) of compound1in CDCl3.Figure S313C NMR spectrum (100 MHz) of compound1in CDCl3.Figure S41H-1H COSY spectrum of compound1in CDCl3.Figure S5HSQC spectrum of compound1in CDCl3.Figure S6HMBC spectrum of compound1in CDCl3.Figure S7NOESY spectrum of compound1in CDCl3.Figure S8HREI (+) MS spectrum of compound1.Figure S9CD spectrum of compound1.Figure S10IR spectrum of compound1.Figure S111H NMR spectrum (400 MHz) of compound2in CDCl3.Figure S1213C NMR spectrum (100 MHz) of compound2in CDCl3.Figure S131H-1H COSY spectrum of compound2in CDCl3.Figure S14HSQC spectrum of compound2in CDCl3.Figure S15HMBC spectrum of compound2in CDCl3.Figure S16NOESY spectrum of compound2in CDCl3.Figure S17HREI (+) MS spectrum of compound2.Figure S18CD spectrum of compound2.Figure S19IR spectrum of compound2.Figure S201HNMR spectrum (400 MHz) of compound3in CDCl3.Figure S2113C NMR spectrum (100 MHz) of compound3in CDCl3.Figure S221H-1H COSY spectrum of compound3in CDCl3.Figure S23HSQC spectrum of compound3in CDCl3.Figure S24HMBC spectrum of compound3in CDCl3.Figure S25NOESY spectrum of compound3in CDCl3.Figure S26HREI(+) MS spectrum of compound3.Figure S27CD spectrum of compound3.Figure S28IR spectrum of compound3.Figure S291H NMR spectrum(400 MHz) of compound4in CDCl3.Figure S3013C NMR spectrum (100 MHz) of compound4in CDCl3.Figure S311H-1H COSY spectrum of compound4in CDCl3.Figure S32HSQC spectrum of compound4in CDCl3.Figure S33HMBC spectrum of compound4in CDCl3.Figure S34NOESY spectrum of compound4in CDCl3.Figure S35HREI (+) MS spectrum of compound4.Figure S36CD spectrum of compound4.Figure S37IR spectrum of compound4.
Acknowledgements
S.T.expressed gratitude for the financial assistance provided by the Thailand Science Research and Innovation (TSRI),Chulabhorn Research Institute (Grant No.36824/4274394 and 36827/4274406),and the Center of Excellence on Environmental Health and Toxicology (EHT),OPS,Ministry of Higher Education,Science,Research and Innovation.The authors acknowledge the involvement of C.Theppitak of Thammasat University in the single-crystal X-ray diffraction investigation.The authors also extend their thanks to P.Kanjanasirirat and K.Jearawuttanakul of the Excellent Center for Drug Discovery (ECDD),Mahidol University,for conducting anti-inflammation tests.
Author contributions
OJ isolation and purification of isolated compounds;JB project planning and coordination,data analysis,and manuscript preparation;ST project planning and coordination,conceptual and technical guidance,compound identification,data analysis,and manuscript preparation;HP compound identification;PB CD and DP4+calculation;SC anti-inflammatory activity assessment;SRuchisansakul plant collection and identification;KC X-ray crystallography analysis;JS molecular docking study;CM and SRuchirawat project supervision.All authors read and approved the final manuscript.
Funding
Thailand Science Research and Innovation,36824/4274394,Sanit Thongnest,36827/4274406,Sanit Thongnest.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its additional information files.
Declarations
Ethics approval and consent to participate
This article does not contain any studies with human participants or animals performed by any of the authors.
Competing interests
The authors declare no conflict of interest associated with this.
Author details
1Laboratory of Natural Products,Chulabhorn Research Institute,Bangkok,Thailand.2Laboratory of Medicinal Chemistry,Chulabhorn Research Institute,Bangkok,Thailand.3Program in Chemical Sciences,Chulabhorn Graduate Institute,Chulabhorn Royal Academy,Bangkok,Thailand.4Center of Excellence on Environmental Health and Toxicology (EHT),OPS,MHESI,Bangkok,Thailand.5Excellent Center for Drug Discovery (ECDD),School of Bioinnovation and Bio-Based Product Intelligence,and Center for Neuroscience,Faculty of Science,Mahidol University,Bangkok,Thailand.6Department of Plant Science,Faculty of Science,Mahidol University,Bangkok,Thailand.7Thammasat University Research Unit in Multifunctional Crystalline Materials and Applications (TU-MCMA),Faculty of Science and Technology,Thammasat University,Pathum Thani,Thailand.8Department of Chemistry,Faculty of Science,Silpakorn University,Nakhon Pathom,Thailand.
Received:6 October 2023
Accepted:19 November 2023
杂志排行

Natural Products and Bioprospecting的其它文章
- A recent update on development,synthesis methods,properties and application of natural products derived carbon dots
- l-Palmitoylcarnitine potentiates plasmin and tPA to inhibit thrombosis
- Ginsenoside compound-K attenuates OVX-induced osteoporosis via the suppression of RANKL-induced osteoclastogenesis and oxidative stress
- The alkynyl-containing compounds from mushrooms and their biological activities
- A comprehensive review on the chemical constituents,sesquiterpenoid biosynthesis and biological activities of Sarcandra glabra
- Natural product rhynchophylline prevents stress-induced hair graying by preserving melanocyte stem cells via the β2 adrenergic pathway suppression