水飞蓟宾对索拉非尼在大鼠体内药动学的影响及机制研究
2023-11-08刘洪涛黄志云张红楠赵媛媛
刘洪涛,张 磊,黄志云,张红楠,张 帆,赵媛媛
水飞蓟宾对索拉非尼在大鼠体内药动学的影响及机制研究
刘洪涛,张 磊,黄志云,张红楠,张 帆,赵媛媛*
河北医科大学第一医院药剂科,河北 石家庄 050000
研究不同剂量水飞蓟宾对索拉非尼在大鼠体内药动学的影响并探究相关机制。雄性SD大鼠随机分为索拉非尼(100 mg/kg)组、低剂量(50 mg/kg)水飞蓟宾+索拉非尼(100 mg/kg)组和高剂量(100 mg/kg)水飞蓟宾+索拉非尼(100 mg/kg)组,每组6只,连续8 d ig空白溶剂或水飞蓟宾后ig索拉非尼,于不同时间点采集血样,测定血浆索拉非尼质量浓度。采用qRT-PCR检测大鼠肝组织中细胞色素P450 3A1(cytochrome P450 3A1,)、尿苷二磷酸葡萄糖醛酸转移酶1A7(UDP-glucuronosyltransferase 1A7,)和小肠组织中P-糖蛋白(P-glycoprotein,)和乳腺癌耐药蛋白(breast cancer resistance protein,)mRNA表达。50 mg/kg水飞蓟宾使索拉非尼的max、AUC0~和AUC0~∞分别增加了47.4%、57.1%和64.7%,100 mg/kg水飞蓟宾使索拉非尼的max、AUC0~和AUC0~∞分别增加了47.6%、80.5%和79.8%;联合给药组小肠组织中和mRNA表达明显受到抑制(<0.05),但肝组织中和mRNA表达没有变化。水飞蓟宾和索拉非尼联用存在药动学相互作用,可能会增加索拉非尼不良反应发生风险,临床联合使用时应加强监测,必要时调整给药剂量。
索拉非尼;水飞蓟宾;药物相互作用;P-糖蛋白;乳腺癌耐药蛋白
肝细胞癌是原发性肝癌最主要的病理分型,占原发性肝癌的75%~85%,因其起病隐匿、恶性程度高且发展迅速,患者在确诊时多处于肝细胞癌晚期,对于该类患者推荐使用系统治疗[1]。索拉非尼是最早批准用于肝细胞癌系统治疗的小分子靶向药物,是肝细胞癌晚期治疗的重要一线药物,但其较高的不良反应发生率,给临床使用带来挑战,积极有效的不良反应治疗和管理也成为了索拉非尼治疗的重要环节。研究显示,约17%的肝癌患者在使用索拉非尼治疗时会出现转氨酶升高[2],加之肝癌患者在自然病程中也可能会伴随肝功能异常,及时适当地使用保肝药物可提高治疗安全性[1],索拉非尼与保肝药物在临床存在广泛联合使用的现象,但是索拉非尼与保肝药物相互作用研究较少。
水飞蓟宾是临床治疗肝功能异常的常用药物之一,具有稳定肝细胞膜、保护肝细胞酶系统、清除肝细胞内活性氧自由基的作用,临床常用于急慢性肝炎的治疗,其在肿瘤患者的应用较为普遍[3]。研究显示,水飞蓟宾可以抑制细胞色素P450 3A(cytochrome P450 3A,CYP3A)等I相代谢酶、尿苷二磷酸葡萄糖醛酸转移酶1A1(UDP-glucuronosyltransferase 1A1,UGT1A1)等II相代谢酶以及P-糖蛋白(P-glycoprotein,P-gp)转运体的活性,能够影响由这些酶代谢或者转运的药物的药动学[4]。索拉非尼在肝脏主要经过UGT1A9和CYP3A4代谢,同时又是P-gp和乳腺癌耐药蛋白(breast cancer resistance protein,BCRP)底物,其与多种药物存在相互作用[5-8]。综上,水飞蓟宾与索拉非尼合用可能发生基于代谢酶和/或转运体的药物相互作用,然而目前尚没有相关研究的报道。因此,开展水飞蓟宾与索拉非尼药动学相互作用研究并初步探究其机制,对于两药物在临床的安全合理使用有重要意义。
1 材料
1.1 动物
SPF级雄性SD大鼠,体质量220~250 g,由北京维通利华实验动物技术有限公司提供,动物生产许可证号SCXK(京)2016-0006。大鼠在标准环境中饲养,实验前12 h禁食,自由饮水。本研究获得河北医科大学第一医院动物伦理委员会批准(No.20230226)
1.2 药品与试剂
索拉非尼对照品(质量分数为99.8%,批号ZZS-20-638-G3)、内标d3-索拉非尼(质量分数为99.9%,批号ZZS-20-X261-A1)均购自上海甄准生物科技有限公司;水飞蓟宾胶囊(批号250704032)购自药店天士力制药集团有限公司;乙腈(批号F23N14201)、甲酸(批号C12531069)为色谱纯,水为娃哈哈纯净水;TRNzol Universal总RNA提取试剂(批号X084)、FastKing cDNA第一链合成试剂盒(批号W0008)、SuperReal PreMix Plus(SYBR Green)荧光定量预混试剂增强版(批号X0829)购自北京天根生化科技有限公司;引物由武汉赛维尔生物科技有限公司合成。
1.3 仪器
LC-30A型超高效液相色谱仪(日本岛津公司);Sciex Triple Quad 5500型串联三重四极杆质谱仪,配有Turbo VTM型电喷雾离子化源(美国AB公司);SLAN-96S型qRT-PCR仪(上海宏石医疗科技有限公司);全自动样品冷冻研磨仪(上海净信实业发展有限公司);Epoch全波长酶标仪(美国Bio-Tek公司);−80 ℃超低温冰箱(美国Thermo Fisher Scientific公司)。
2 方法
2.1 大鼠体内药动学研究
18只雄性SD大鼠,随机分为索拉非尼(100 mg/kg,相当于临床剂量14 mg/kg)组、低剂量(50 mg/kg,相当于临床剂量7 mg/kg)水飞蓟宾+索拉非尼(100 mg/kg)组和高剂量(100 mg/kg,相当于临床剂量14 mg/kg)水飞蓟宾+索拉非尼(100 mg/kg)组,每组6只,实验前禁食12 h,自由饮水。所有药物以0.5%羧甲基纤维素钠混悬,大鼠连续8 d ig水飞蓟宾或者空白溶剂后,ig索拉非尼,在给药前和给药后1、3、5、6、7、8、10、12、24、36、48、72、96 h取血0.3 mL于肝素化抗凝管中,血液样本4500 r/min离心10 min,取血浆保存于−80 ℃冰箱待分析。
2.2 水飞蓟宾对大鼠肝脏CYP3A1、UGT1A7和小肠组织P-gp、BCRPmRNA表达的影响
采血结束后,大鼠ip 2%戊巴比妥麻醉后,迅速取出肝脏及小肠组织,组织经预冷的生理盐水冲洗,滤纸吸湿,放入组织包埋盒,在液氮中速冻后保存在−80 ℃待分析。使用总RNA快速抽提试剂盒提取肝脏和小肠组织中的总RNA,测定总RNA的浓度,根据260 nm与280 nm的吸光度()比值评估总RNA的纯度,该值在1.8~2.0。使用FastKing cDNA第一链合成试剂盒将总RNA逆转录成cDNA。采用SuperReal PreMix Plus(SYBR Green)试剂盒进行定量PCR反应。反应程序设置预变性95 ℃持续15 min,PCR反应(40个循环,95 ℃、10 s,60 ℃、32 s),引物序列见表1。
2.3 溶液的配制
精密称取索拉非尼对照品适量,用二甲基亚砜溶液溶解,制成终质量浓度为1 mg/mL的对照品储备液。取适量对照品储备液,用50%乙腈稀释为50、150、500、2000、8000、2×104、5×104ng/mL的混合标准曲线工作溶液。同法配制低、中、高混合质控工作溶液和稀释可靠性工作溶液,索拉非尼质量浓度分别为100、5000、3.75×104、1×105ng/mL。

表1 引物序列
称取d3-索拉非尼1 mg于1 mL二甲基亚砜中溶解,制成1 mg/mL的内标储备液,用50%乙腈稀释得到500 ng/mL的混合内标工作溶液。分析前,所有溶液保存于−20 ℃冰箱。
2.4 血浆样品处理
血浆样品50 μL,加入内标工作溶液5 μL,加入乙腈150 μL,涡旋混合1 min,12 000 r/min离心10 min,取上清液100 μL加入200 μL 50%乙腈,涡旋混匀,转移至进样小瓶,待进样分析。
2.5 LC-MS/MS条件
2.5.1 色谱条件 ZORBAX SB-C18色谱柱(100 mm×2.1 mm,3.5 µm),流动相为乙腈(A)-含0.1%甲酸的5 mmol/L乙酸铵溶液(B),梯度洗脱:0~2.5 min,60% B;2.5~3.5 min,60%~90% B;3.5~5.5 min,90% B;5.5~5.6 min,90%~60% B;5.6~6.6 min,60% B。体积流量为0.7 mL/min;进样体积为5 μL。
2.5.2 质谱条件 电喷雾离子源正离子检测模式,多反应监测扫描,索拉非尼和内标d3-索拉非尼定量离子对分别为/465.2→270.2、468.2→255.4;去簇电压140 V;碰撞能量45 eV;源喷射电压5500 V;雾化气压力413.7 kPa;加热气压力344.75 kPa;气帘气压力137.9 kPa;离子源温度500 ℃。
2.6 方法学考察
2.6.1 选择性 取6份不同来源的空白血浆以及制备的定量限校正标样,按照“2.4”项下方法处理后获得样品色谱图,考察方法的选择性。
2.6.2 标准曲线和定量限 取“2.3”项下对照品工作溶液5 μL加入45 μL空白血浆,涡旋混合,制成索拉非尼质量浓度分别为5、15、50、200、800、2000、5000 ng/mL的校正标样,按“2.4”项下方法处理后,进样分析,记录峰面积。以分析物的质量浓度为横坐标,分析物与内标的峰面积比为纵坐标,采用最小加权二乘法以1/2为权重因子进行曲线拟合得回归方程。
2.6.3 精密度和准确度 配制含索拉非尼质量浓度为5.0、10.0、500.0、3 750.0 ng/mL的定量下限、低、中、高4个质量浓度的质控血浆样品,按“2.4”项下方法处理,每个质量浓度平行6份,连续3 d测定,计算日内、日间精密度和准确度。
2.6.4 基质效应 取6份不同来源的空白血浆50 μL,按“2.4”项下方法处理,不加内标,取上清液即为空白基质溶液,向空白基质加入低、高质量浓度的质控工作溶液和内标溶液适量(使其质量浓度与低、高质控样本质量浓度相同),每个质量浓度6样本,测得峰面积记为B;以50%乙腈代替空白基质其余操作同上,测得峰面积记为C,以B/C分别计算分析物和内标的基质效应因子,再以分析物的基质效应因子除以内标的基质效应因子计算归一化的基质效应因子。
2.6.5 提取回收率 配制低、中、高质量浓度的质控样品,按“2.4”项下方法处理,进样分析后,记录分析物与内标的峰面积比值为A;取空白血浆,同法处理后加入3个质量浓度质控工作溶液和内标溶液,使之与质控样品质量浓度相同,记录分析物与内标的峰面积比值为B,A/B即为分析物提取回收率。
2.6.6 稳定性 配制低、高质量浓度质控样品分别考察含药血浆在室温下放置8 h、2~8 ℃冰箱放置24 h、−80 ℃放置14 d、处理后样品在自动进样器放置12 h、−80 ℃冻融3次的稳定性。
2.6.7 稀释可靠性 制备2倍于定量上限质量浓度的血浆样本,再用空白血浆稀释该样品5倍,平行6份,测定结果与标示量的偏差。
2.6.8 残留效应 定量上限质量浓度后进样空白基质生物样本,考察本方法的残留效应。
2.7 统计学分析

3 结果
3.1 方法学验证
3.1.1 选择性 索拉非尼及内标的保留时间分别为1.17、1.16 min。空白血浆中的内源性物质不干扰分析物的测定,方法的选择性良好,色谱图见图1。
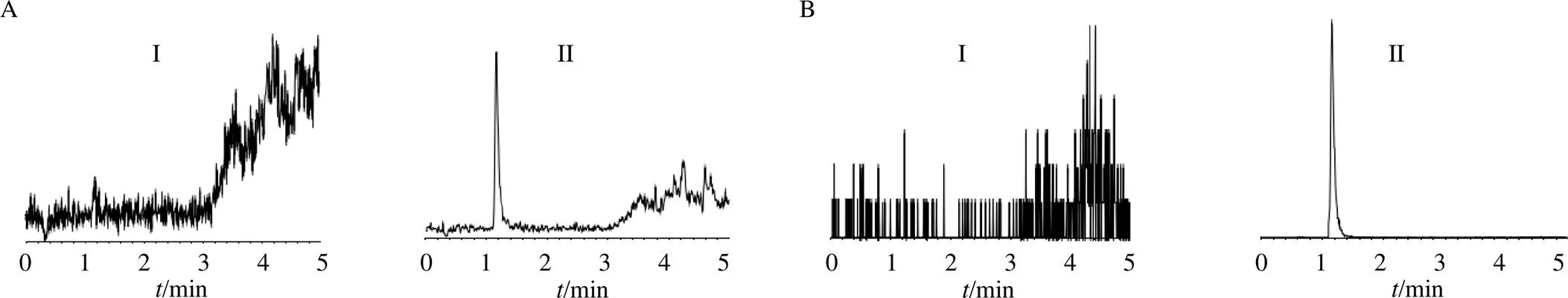
I-空白血浆色谱图 II-加入5 ng/mL索拉非尼和内标的空白血浆
3.1.2 标准曲线及定量限 索拉非尼在5~5000 ng/mL内线性关系良好,标准曲线为=4.28×10−3-2.46×10−4(=0.996 3),连续测定6份定量限样本,其精密度与准确度均满足要求,定量限为5 ng/mL。
3.1.3 精密度和准确度 定量限、低、中、高质量浓度质控样品的日内精密度、日间精密度及准确度均满足要求(表2)。
3.1.4 基质效应和提取回收率 索拉非尼低、高质量浓度质控样品经内标校正的基质效应分别为97.0%和100.6%,RSD分别为7.0%和4.0%,结果表明基质的存在不干扰索拉非尼的测定。索拉非尼的提取回收率为95.5~101.8%(表3)。
表2 精密度和准确度(, n = 6)
表3 基质效应和提取回收率(, n = 6)
3.1.5 稳定性 索拉非尼含药血浆在室温放置8 h、2~8 ℃冰箱放置24 h、−80 ℃放置14 d、处理后样品在自动进样器放置12 h、−80 ℃冻融3次的稳定性均未受到影响,其准确度为96.4%~107.2%,RSD均小于10%(表4),表明在上述条件下,血浆中索拉非尼稳定性良好。
3.1.6 稀释可靠性 索拉非尼稀释5倍的样品测定结果与标示量偏差为3.2%,RSD小于15%,索拉非尼血浆药物质量浓度高于定量上限时,可以采用空白血浆稀释后准确测定。
3.1.7 残留效应 最高浓度校正标样后连续分析的1个空白基质样品在分析物和内标保留时间处无明显干扰峰,说明高质量浓度样品对低质量浓度样品的测定无残留影响。
3.2 大鼠体内药动学结果
索拉非尼单独给药和联合水飞蓟宾后索拉非尼的血药浓度-时间曲线见图2,索拉非尼的主要药动学参数见表5。当索拉非尼与低、高剂量水飞蓟宾联合给药时,索拉非尼的AUC0~t、AUC0~∞和max明显增加(<0.05),表观分布容积()和清除率(CL)降低。低剂量水飞蓟宾联合索拉非尼组和高剂量水飞蓟宾联合索拉非尼组的药动学参数差异无统计学意义。低剂量水飞蓟宾组和索拉非尼单用组的药动学参数max和AUC0~t几何均值比的90% CI分别为128.04%~174.09%和120.31%~233.67%,高剂量水飞蓟宾组和索拉非尼单用组的药动学参数max和AUC0~t几何均值比的90% CI分别为115.49%~184.63%和127.13%~274.34%,均不在80.00%~125.00%,结果表明低、高剂量水飞蓟宾均明显影响大鼠体内索拉非尼的药动学过程。低剂量水飞蓟宾使索拉非尼的AUC0~t升高1.67倍,高剂量水飞蓟宾使索拉非尼的AUC0~t升高1.87倍,结果均大于1.25倍且小于2倍,表明水飞蓟宾对大鼠体内索拉非尼体内过程可能存在弱抑制。
表4 稳定性(, n = 6)
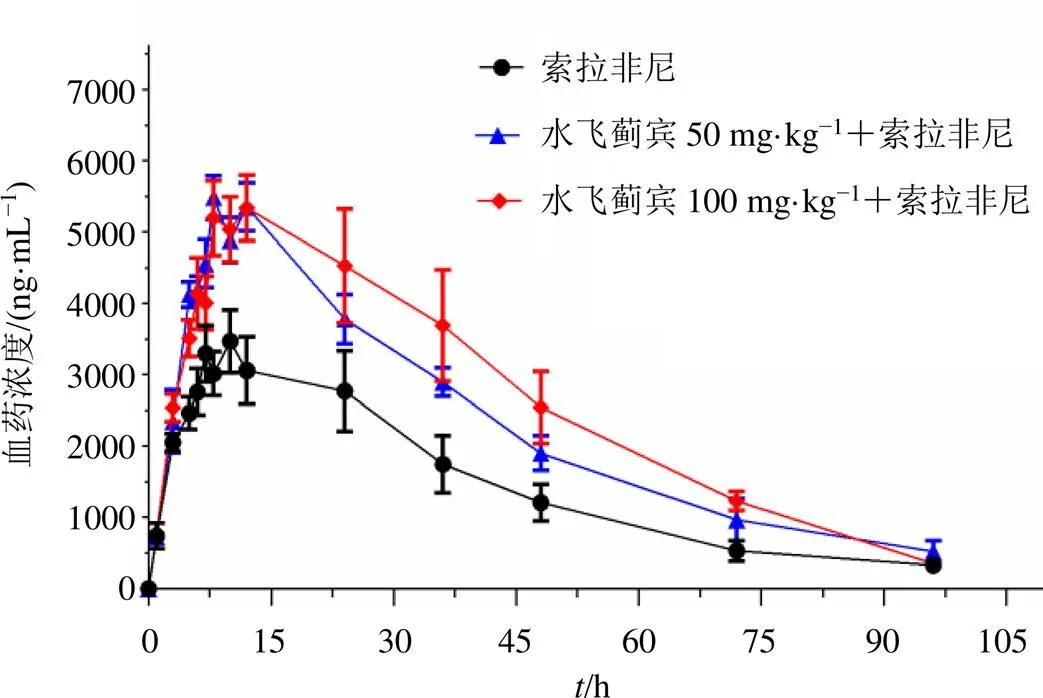
图2 索拉非尼在大鼠体内的平均药-时曲线(, n = 6)
表5 索拉非尼的药动学参数(, n = 6)
与索拉非尼组比较:*<0.05
*< 0.05sorafenib group
3.3 水飞蓟宾对大鼠肝脏CYP3A1、UGT1A7和小肠组织P-gp、BCRPmRNA表达的影响
如图3所示,与索拉非尼组比较,低、高剂量水飞蓟宾联合索拉非尼组小肠组织中和mRNA表达水平均明显降低(<0.05),肝脏组织中和mRNA表达无明显变化。
4 讨论
索拉非尼因其较优异的抗肿瘤效果,一直是晚期肝癌患者的重要治疗手段,但因肿瘤患者常存在共患病,索拉非尼与其他药物联合使用的现象十分普遍,加之索拉非尼的体内过程容易受到代谢酶和转运体活性改变的影响[9-10],如合用药物影响代谢酶和转运体,药物相互作用的发生风险也会增加。药物相互作用发生隐匿,通常对于没有治疗药物监测的药物使用中很难发觉,而一旦发现药物相互作用,一般是患者已经出现了药物不良反应,因此,主动探究联用药物之间相互作用发生的可能,对于索拉非尼的安全合理使用十分必要。肝癌患者在自然病程中或治疗过程中常会伴随肝功能异常,及时适当地使用保肝药物可以降低并发症和改善生活质量,临床上水飞蓟宾因其较好的保肝效果也是晚期肝癌患者的常用药物之一,其与索拉非尼联合使用的现象也十分普遍。本研究在大鼠体内,从整体水平考察了不同剂量多次给予水飞蓟宾与单次给予索拉非尼的药动学相互作用,结果发现50 mg/kg水飞蓟宾使索拉非尼的max、AUC0~t和AUC0~∞分别增加了47.4%、57.1%和64.7%,100 mg/kg水飞蓟宾使索拉非尼的max、AUC0~t和AUC0~∞分别增加了47.6%、80.5%和79.8%,鉴于索拉非尼达稳态后药物暴露有2.5~7.0倍的蓄积,因此两药稳态下合用后药物暴露增加程度可能加大。研究显示索拉非尼体内暴露量与不良反应相关[11],药物暴露增加会导致手足综合征和腹泻的发生风险增加,不利于药物治疗的安全性。因此当临床上水飞蓟宾与索拉非尼联合使用时,需要临床给予重视,对索拉非尼的疗效和安全性应加强监测,必要时减少索拉非尼的给药剂量或者进行治疗药物监测,以免影响药物治疗结局。
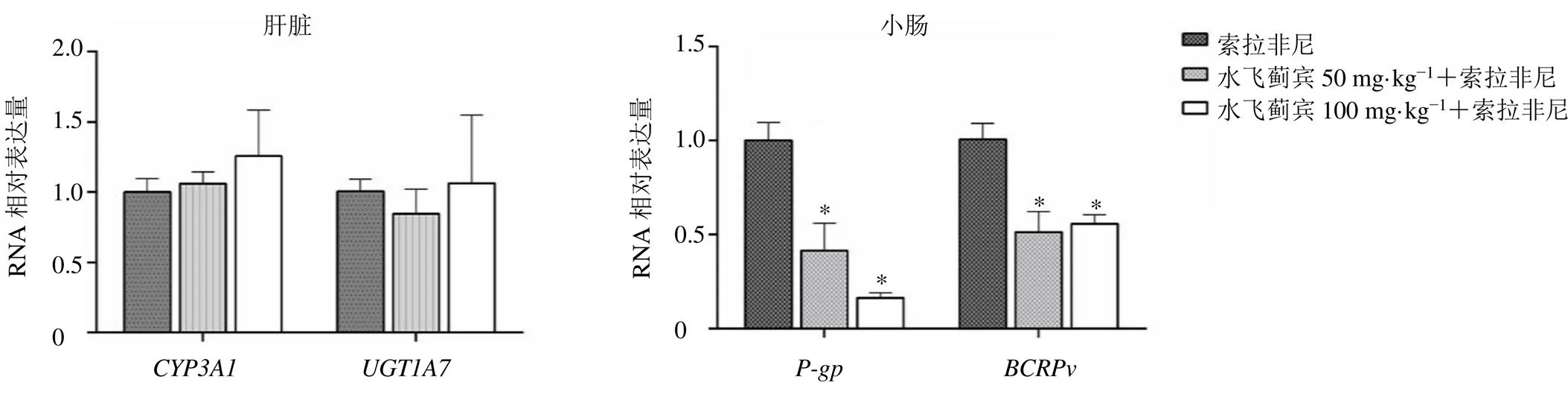
与索拉非尼组比较:*P<0.05
为进一步探究水飞蓟宾影响索拉非尼体内药物暴露的原因,本研究基于既往研究[12-13]选择了与索拉非尼相关的药物代谢酶(CYP3A4、UGT1A9)和转运体(P-gp、BCRP)作为靶点,通过相关基因表达情况初步探究水飞蓟宾改变索拉非尼体内过程的原因。UGT1A9和CYP3A4是索拉非尼在人体的主要代谢酶,在大鼠体内两者的功能分别由UGT1A7和CYP3A1所取代,因此本研究以UGT1A7和CYP3A1作为研究靶点。尽管多项体外研究显示水飞蓟宾是UGTs和CYP3A4的抑制剂[14-15],但是本研究没有得到相同结果,可能是因为水飞蓟宾的生物利用度低,在肝脏没有达到体外研究的药物浓度。外排转运体在索拉非尼吸收中起着重要作用,索拉非尼生物利用度较低,可能与肠道外排相关[13],体外研究显示索拉非尼为P-gp和BCRP底物,其对BCRP的亲和力强于P-gp[16]。水飞蓟宾是BCRP底物,对P-gp的影响存在相反结果,这可能与给药剂量、给药频次有关[4]。本研究显示大鼠连续8 d ig给予50、100 mg/kg水飞蓟宾,均可以抑制大鼠小肠和的mRNA表达,两组间没有差异,推测可能是因为水飞蓟宾生物利用度极低(约0.95%)[17],即使增加剂量其吸收进入小肠细胞的药物并未增加多少,导致其对转运体产生的作用没有差别。综上所述,水飞蓟宾可能是通过抑制大鼠小肠P-gp和BCRP而使索拉非尼生物利用度增加,两者也可能同时竞争小肠BCRP而使索拉非尼的生物利用度增加,具体机制还有待进一步研究。本研究为动物体内开展的药动学研究,由于物种的差异,并不能完全代表人体体内情况。索拉非尼与水飞蓟宾稳态下长期药动学相互作用及药效学相互作用尚需进一步研究。
利益冲突 所有作者均声明不存在利益冲突
[1]中华人民共和国国家卫生健康委员会. 原发性肝癌诊疗指南 (2022年版) [J]. 肿瘤综合治疗电子杂志, 2022, 8(2): 16-53.
[2]Kudo M, Finn R S, Qin S K,. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial [J]., 2018, 391(10126): 1163-1173.
[3]张志鹏, 孟艳秋, 王趱. 水飞蓟宾及其衍生物生物活性及作用机制的研究进展[J]. 中草药, 2021, 52(12): 3717-3724.
[4]Xie Y, Zhang D Q, Zhang J,. Metabolism, transport and drug-drug interactions of silymarin [J]., 2019, 24(20): 3693.
[5]Mross K, Steinbild S, Baas F,. Results from anand a clinical/pharmacological phase I study with the combination irinotecan and sorafenib [J]., 2007, 43(1): 55-63.
[6]Karbownik A, Sobańska K, Grabowski T,.assessment of the drug interaction between sorafenib and paracetamol in rats [J]., 2020, 85(6): 1039-1048.
[7]Karbownik A, Szkutnik-Fiedler D, Grabowski T,. Pharmacokinetic drug interaction study of sorafenib and morphine in rats [J]., 2021, 13(12): 2172.
[8]Karbownik A, Szkutnik-Fiedler D, Czyrski A,. Pharmacokinetic interaction between sorafenib and atorvastatin, and sorafenib and metformin in rats [J]., 2020, 12(7): 600.
[9]Karbownik A, Miedziaszczyk M, Grabowski T,.assessment of potential for UGT-inhibition-based drug-drug interaction between sorafenib and tapentadol [J]., 2020, 130: 110530.
[10]Bins S, van Doorn L, Phelps M A,. Influence of OATP1B1 function on the disposition of sorafenib-β--glucuronide [J]., 2017, 10(4): 271-279.
[11]Boudou-Rouquette P, Ropert S, Mir O,. Variability of sorafenib toxicity and exposure over time: A pharmacokinetic/pharmacodynamic analysis [J]., 2012, 17(9): 1204-1212.
[12]Järvinen E, Deng F, Kiander W,. The role of uptake and efflux transporters in the disposition of glucuronide and sulfate conjugates [J]., 2021, 12: 802539.
[13]Tandia M, Mhiri A, Paule B,. Correlation between clinical response to sorafenib in hepatocellular carcinoma treatment and polymorphisms of P-glycoprotein (ABCB1) and of breast cancer resistance protein (ABCG2): Monocentric study [J]., 2017, 79(4): 759-766.
[14]Sridar C, Goosen T C, Kent U M,. Silybin inactivates cytochromes P450 3A4 and 2C9 and inhibits major hepatic glucuronosyltransferases [J]., 2004, 32(6): 587-594.
[15]Beckmann-Knopp S, Rietbrock S, Weyhenmeyer R,. Inhibitory effects of silibinin on cytochrome P-450 enzymes in human liver microsomes [J]., 2000, 86(6): 250-256.
[16]Agarwal S, Sane R, Ohlfest J R,. The role of the breast cancer resistance protein (ABCG2) in the distribution of sorafenib to the brain [J]., 2011, 336(1): 223-233.
[17]Tvrdý V, Pourová J, Jirkovský E, Křen V, Valentová K, Mladěnka P. Systematic review of pharmacokinetics and potential pharmacokinetic interactions of flavonolignans from silymarin [J]., 2021, 41(4): 2195-2246.
Effect and mechanism of silibinin on pharmacokinetics of sorafenib in rats
LIU Hong-tao, ZHANG Lei, HUANG Zhi-yun, ZHANG Hong-nan, ZHANG Fan, ZHAO Yuan-yuan
Department of Pharmacy, The First Hospital of Hebei Medical University, Shijiazhuang 050000, China
To investigate the effect of silibinin on pharmacokinetics and mechanism of sorafenib in rats.Male SD rats were randomly divided into sorafenib (100 mg/kg) group, low-dose silibinin (50 mg/kg) + sorafenib (100 mg/kg) group and high-dose silibinin (100 mg/kg) + sorafenib (100 mg/kg) group, with six rats in each group. After 8 d of ig blank solvent or silibinin, rats were ig sorafenib, blood samples were collected at different time points to determine the concentration of sorafenib in plasma. The mRNA expressions of cytochrome P450 3A1 (CYP3A1), UDP-glucuronosyltransferase 1A7 (UGT1A7) in liver tissue and P-glycoprotein (P-gp), breast cancer resistance protein (BCRP) in small intestine tissue of rats were detected by qRT-PCR.Silibinin at dose of 50 mg/kg increased themax, AUC0−tand AUC0−∞of sorafenib by 47.4%, 57.1% and 64.7% respectively, while 100 mg/kg silibinin increased themax, AUC0−tand AUC0−∞of sorafenib by 47.6%, 80.5% and 77.5% respectively. The expressions ofandmRNA in small intestine were obviously inhibited (< 0.05), but the expressions ofandmRNA in liver tissue had no change.There is a pharmacokinetic interaction between silibinin and sorafenib, which may increase the risk of adverse reactions of sorafenib. Monitoring should be strengthened in clinical combined use, and the dosage should be adjusted if necessary.
sorafenib; silibinin; drug-drug interaction; P-glycoprotein; breast cancer resistance protein
R285.62
A
0253 - 2670(2023)21 - 7104 - 07
10.7501/j.issn.0253-2670.2023.21.019
2023-06-11
河北医科大学星火计划课题(XHJH202301)
刘洪涛,副主任药师,主要从事医院药学相关研究。E-mail: lhtyl16@126.com
通信作者:赵媛媛,主任医师,主要从事精神疾病及药物研究。E-mail: zhaoyuanyuan9955@163.com
[责任编辑 李亚楠]
