基于cfDNA的NGS技术探究术后脓毒症患者血液微生物群特征
2023-11-07王腾蛟
梁 栋 梅 月 王腾蛟 喻 东
(中国人民解放军海军军医大学转化医学研究中心,上海 200433)
随着外科无菌术、重症监护技术的进步,以及抗菌药物使用的规范化,术后感染得到了极大改善,但脓毒症依然是常见的术后并发症,威胁着患者生命[1]。脓毒症是由微生物病原体侵入血液而引发的全身性炎症反应[2]。细菌感染是诱发脓毒症的首要因素,常见的病原体包括肺炎链球菌、大肠埃希菌、铜绿假单胞菌、金黄色葡萄球菌和克雷伯菌属细菌[3-6],由真菌和病毒引发的脓毒症相对较少[7-8]。目前,诊断脓毒症的金标准是传统的血培养法,但该方法有耗时长、敏感性和特异性较弱等局限[9]。因此,更便捷的诊断工具和高敏感性、高特异性的病原体鉴定技术,成为感染性疾病诊断的迫切需求。
目前,血液和其他体液中生物标志物的检测已广泛用于医学研究,并在临床诊断领域有着潜在的应用前景。在现有的诊断工具中,细胞游离DNA(cell-free DNA,cfDNA)作为一种高效、非侵袭性的生物标志物,已被应用于肿瘤、产前诊断和器官移植排斥等临床实践中[10-12],但其在传染病领域却鲜有应用[13]。近年来,基因组测序技术飞速发展,下一代测序(next-generation sequencing,NGS)技术在感染性疾病的诊断中展现出较大的价值[14]。有研究结果显示,全基因组NGS能够有效识别血浆中的病原体,并在24 h之内完成对脓毒症的诊断[9]。如果将基于cfDNA的NGS技术应用于脓毒症的诊断,有望实现病原微生物的精准、快速识别,以及疾病的非侵袭性诊断。本研究拟通过对公共数据库中cfDNA的测序数据进行再次分析,探索脓毒症患者(sepsis patient,SP)和对照患者(control patient,CP)血浆中微生物组成的差异性,以更好地理解微生物在引发脓毒症中所起的重要作用,帮助临床有效预防脓毒症。
1 材料和方法
1.1 原始测序数据的收集
从SRA-NCBI公共数据库(BioProject:PRJEB21872和PRJEB30958)下载cfDNA原始测序数据。测序样本来源于239例外科手术术后SP和34例外科手术术后未发生感染的CP的血浆样本[15-16]。样本的测序信息见表1。
1.2 数据处理和分析
首先,通过SRA Toolkit软件将原始的sra格式文件转化为Fastq格式,并通过FastQC进行质量控制[17]。然后,用Kraken2软件进行物种分类分析,Kraken2软件的比对数据库包括古菌、细菌、人类和病毒[18-19]。
1.3 统计学方法
采用SPSS 19.0软件进行统计分析。计数资料用例或率表示。正态分布计量资料用±s表示。用线性判别分析效应分析(linear discriminant analysis effect size,LEfSe)评估组间物种丰度的差异性,以物种的线性判别分析(linear discriminant analysis,LDA)值>2,且P<0.05(Kruksal-Wallis检验)为显著性富集。
2 结果
2.1 微生物分布特征
利用Kraken2将DNA序列与参考数据库进行比对,得到每个样本的物种分类信息和相对丰度(表2),结果显示,SP和CP分别有(99.11±5.42)%和(99.84±0.47)%的序列被识别为细菌,(0.89±5.42)%和(0.16±0.47)%的序列被识别为病毒,(0.002 6±0.015 0)%和(0.000 14±0.000 83)%的序列被识别为古生菌。
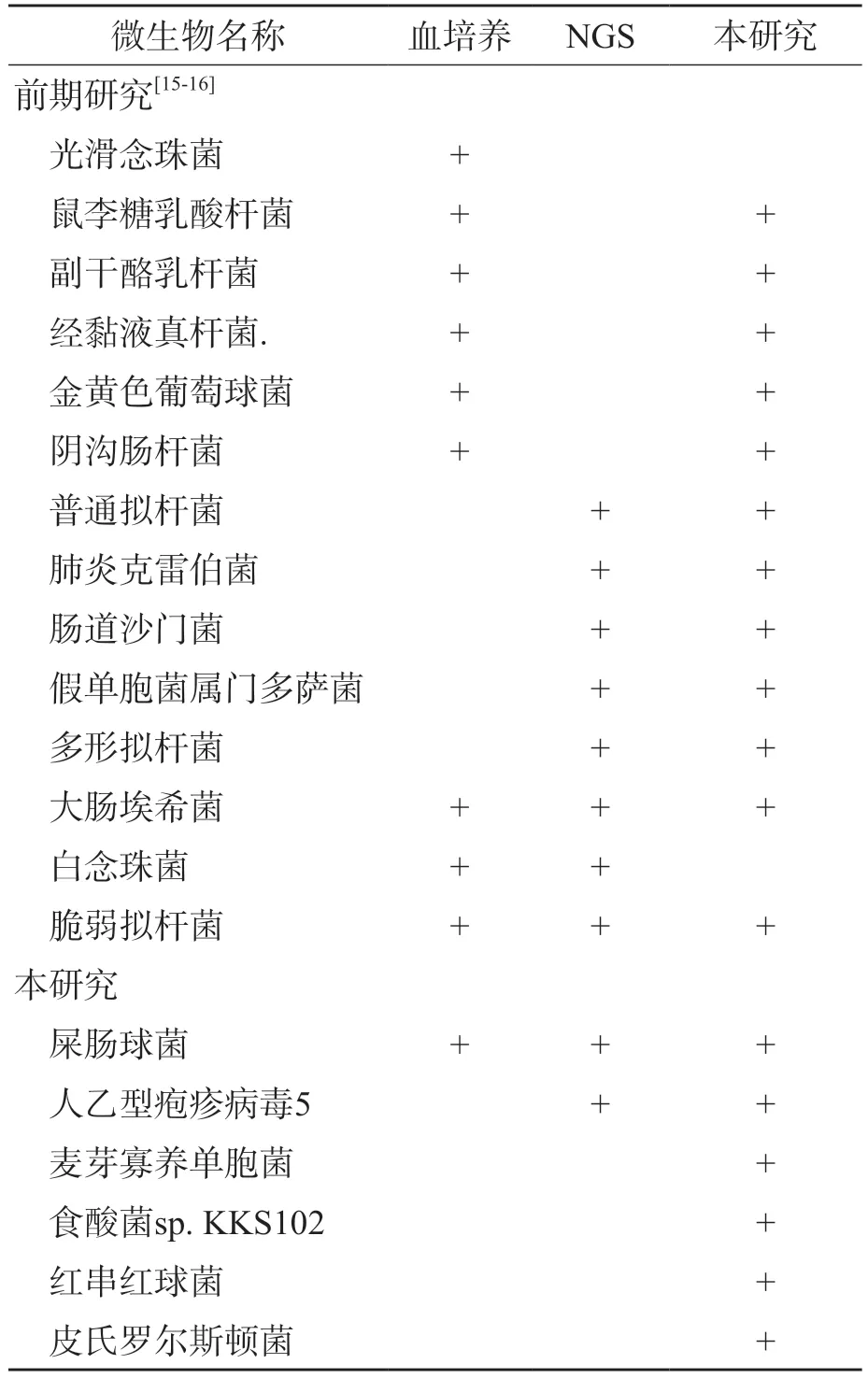
表2 前期研究[15-16]和本研究之间的物种比较
2.2 微生物多样性差异分析
CP和SP血液中分布的微生物在门、纲、目、科、属、种水平上的数量分别为15、25、82、133、267、634和16、37、121、205、432、1 085,见表2。对CP和SP组样本进行α多样性分析和β多样性分析,发现SP样本中微生物的总体多样性显著低于CP样本,见图1。
2.3 CP和SP微生物相对丰度差异分析
在CP中,变形菌门是最主要的物种(95.23%),其次是放线菌门(4.36%),还有0.19%的厚壁菌门和0.01%的拟杆菌门,以及极少量的酸杆菌门、产水菌门、衣原体门、蓝藻门、梭杆菌门、Kiritimatiellaeota、硝化螺旋菌门、浮霉菌门、螺旋体门和疣微菌等。与CP相比,SP血浆样本中放线菌门(28.02%)、厚壁菌门(1.82%)和拟杆菌门(0.61%)显著增多,变形菌门(69.03%)显著减少。见图2。
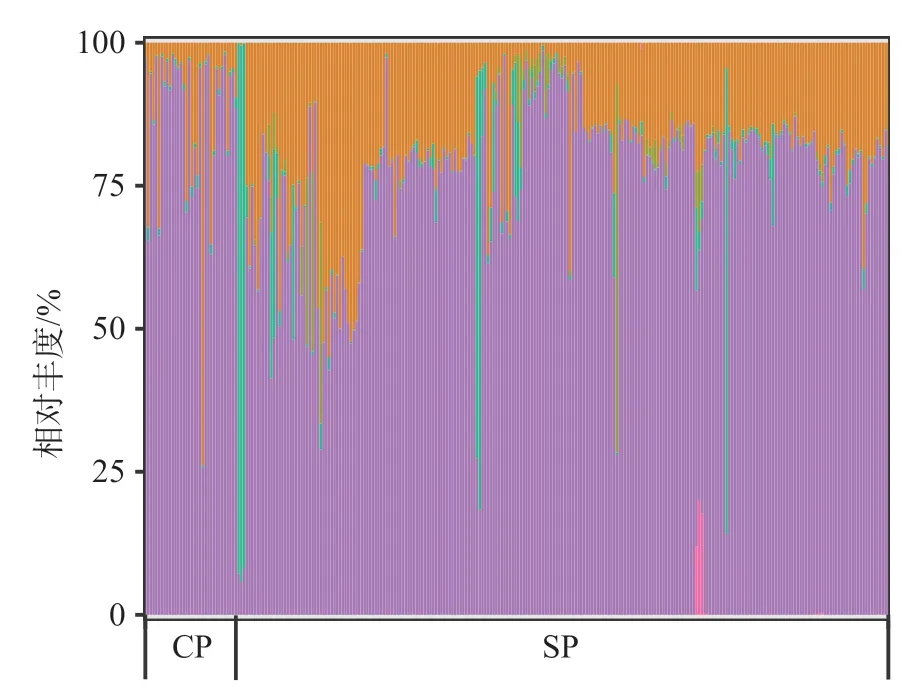
图2 SP和CP物种在门分类学水平上的分布
用LEfSe对SP和CP的微生物群落结构进行比对分析,结果显示,SP和CP样本间差异具有统计学意义的物种数量为260个(LDA>2,P<0.05),其中差异性较大(LDA>4,P<0.05)的物种有25个。SP显著富集的物种数量为14个,包括1门(放线菌门)、1纲(放线菌纲)、2目、4科、2属、4种;CP显著富集的物种数量为11个,包括1门(变形菌门)、1纲(γ变形菌纲)、2目、2科、2属、3种。见图3。

图3 SP和CP微生物差异分析
除细菌外,SP和CP病毒也表现出较大的差异性,包括细小病毒科(CP为0.000 24%,SP为0)、指环病毒科(CP为0.14%,SP为0.002 3%)和有尾噬菌体目(CP为0.027%,SP为0.093%)。
2.4 SP优势菌种的鉴别
在种水平,前20个相对丰度较高的菌种中,有16个在SP和CP之间表现出了显著性差异(图4)。其中,SP有12个菌种(嗜麦芽寡养单胞菌、红串红球菌、食酸菌sp. KKS102、皮氏罗尔斯顿菌、屎肠球菌、人乙型疱疹病毒5、食酸菌sp. JS42、脆弱拟杆菌、大肠埃希菌、Azospira oryzae、中间硫杆菌和多形拟杆菌)显著增多,4个菌种(恶臭假单胞菌、痤疮丙酸杆菌、罗氏假单胞菌和铜绿假单胞菌)显著减少。值得注意的是,屎肠球菌、大肠埃希菌、Azospira oryzae和中间硫杆菌这4种兼性厌氧菌仅存在于SP中。

图4 SP和CP相对丰度居前20位的物种
将SP显著增加,且相对丰度>1%的物种视为代表性物种,选出6种代表性物种,包括麦芽寡养单胞菌、红串红球菌、食酸菌sp. KKS102、皮氏罗尔斯顿菌、屎肠球菌和唯一的病毒人乙型疱疹病毒5。见表1、图4。
3 讨论
本研究识别出6种代表性物种,包括麦芽寡养单胞菌、红串红球菌、食酸菌sp. KKS102、皮氏罗尔斯顿菌、屎肠球菌和人乙型疱疹病毒5。有大量文献提示这几个物种与脓毒症的发生、发展具有潜在的联系。麦芽寡养单胞菌和皮氏罗尔斯顿菌属于变形菌门,屎肠球菌属于厚壁菌门,3种细菌都是医院内常见的条件致病菌[20-22],且是医院内感染的重要致病菌,包括脓毒症[23-25]。值得注意的是,在健康人群中,厚壁菌门和变形菌门常定植于肠道,是肠道菌群中的2类优势菌门[26]。然而,与健康人群相比,危重症患者的肠道中厚壁菌门显著减少,变形菌门显著增加[27]。这种微生物结构的剧烈变动会导致肠道内环境紊乱,进而可能引发肠道细菌移位和肠源性脓毒症[28]。本研究发现,脓毒症患者血液中所占比例最多的菌种是变形菌门(69.03%),而厚壁菌门仅占1.82%。脓毒症患者血液与危重症患者肠道微生物结构的相似性提示肠道细菌发生了移位[29],而细菌移位在脓毒症早期起重要作用[30]。本研究中,麦芽寡养单胞菌、皮氏罗尔斯顿菌和屎肠球菌的定量变化可能是导致肠道屏障功能紊乱的关键因素,继而诱发致病性细菌移位和肠源性脓毒症。
与上述3种条件致病菌不同,红串红球菌和食酸菌sp. KKS102是侵入性病原体[31-32]。红串红球菌虽然广泛分布于自然环境中,但很少有人类感染,目前仅查到2例红串红球菌引发的血液感染病例[31,33]。食酸菌sp. KKS102也很少引发人类感染[34],查阅文献,尚未见除本研究外的食酸菌sp. KKS102引发的脓毒症的报道。红串红球菌和食酸菌sp. KKS102难以检测的原因可能是物种的难以鉴定和生长延迟[33-34]。另外,这2种菌的非嗜热生长特性也是导致其极少引发人类感染的原因之一[31,35]。虽然红串红球菌和食酸菌sp. KKS102极少引发人类感染,但本研究结果提示其依旧是脓毒症的潜在致病菌,不能简单地将其看作环境污染菌而忽视。
人乙型疱疹病毒5是本研究脓毒症患者血液中唯一相对丰度显著增加的病毒。人乙型疱疹病毒5属于巨细胞病毒属(cytomegalovirus,CMV),CMV是一类在免疫缺陷宿主中常见的机会性病原体[36]。细菌性脓毒症是CMV重激活的重要触发因素,且伴随着较差的临床预后,如死亡率增加、重症监护病房停留和住院时间延长,机械通气时间延长,以及二次感染机会增加[36-37]。幸运的是,CMV重激活的风险可被显著降低,由于重激活事件往往出现在发病7~28 d,在发病8 d内监测可将CMV重激活的风险降至1%,甚至是0[38]。
有研究发现,可通过控制感染源来减少红串红球菌、 食酸菌sp. KKS102和人乙型疱疹病毒5的感染风险,包括移除污染设备(如中心静脉导管)和医疗器械的表面消毒处理[33,37,39]。除控制感染源外,对肠道微生物的干预也是应对肠源性脓毒症的有效医疗策略。近年来,相关临床病例报道均提示粪菌移植(fecal microbiota transplantation,FMT)在脓毒症治疗中有积极作用,粪便微生物在患者体内表现出了较好的干预效果[40]。本研究发现的6种代表性微生物可能是引发术后脓毒症患者感染的潜在病原体。然而,值得注意的是,这6种潜在病原体与引发脓毒症的常见病原体(肺炎链球菌、大肠埃希菌、铜绿假单胞菌、金黄色葡萄球菌和克雷伯菌属细菌)有较大差异,可能是由以下3方面因素造成的:1)不同的生物信息学分析方法所比对的微生物参考数据库不一致;2)NCBI测序数据可能有偏差,不同实验室的研究对象具有一定的选择性,导致常见微生物测序比例大大降低,而部分罕见微生物比例则有所增加;3)术后脓毒症患者的致病菌受医院环境影响较大,不同医院流行病原体不同。因此,本研究结果主要为脓毒症的诊断和治疗提供数据参考,同时提示部分罕见微生物虽较少引发人类感染,但不能忽视其潜在的致病性。
本研究基于公开的基因组测序数据,但在研究目的和方法上与之前的研究[15-16]完全不同。在之前的研究中,通过与非感染对照组的比对,计算基于NGS测序结果的脓毒症量化指示分数(sepsis indicating quantifier score,SIQ),对单个样本检测到的每个微生物进行定量、概率性评估[16],分析脓毒症患者的病原体感染状态,以鉴定NGS诊断的适用性,并将评估结果与传统的血培养法进行直接比较[15],着重关注单个脓毒症患者体内病原微生物的识别,没有进一步分析脓毒症患者体内富集的微生物。本研究可视为对之前研究的补充,关注SP和CP微生物的总体差异性,并对一些重要的差异性物种进行了详细的分析。另外,从结果上比较,之前的研究[15-16]通过血培养或NGS检测出的菌种在本研究中基本都被检测了出来(表2)。
本研究也有一定的局限性,样本数量相对较少,临床信息不够完整,限制了临床特征与微生物群落的相关性分析,未进行相关的实验性验证。在之后的研究中,我们将努力突破局限,进一步完善相关微生物与脓毒症发生、发展之间的关联性。
综上,本研究基于较大的人群队列基因组数据,以基于血浆cfDNA的NGS技术,初步探索了术后感染和未感染患者血液微生物组成的差异性,为进一步理解微生物与脓毒症之间的联系提供了数据支撑。本研究揭示了脓毒症患者血液中微生物多样性的显著下调,以及微生物群落结构的明显变化,发现了6种可能在脓毒症发生、发展过程中发挥重要作用的代表性微生物。
