自噬对肝衰竭的保护作用机制与临床价值
2023-11-06胡洋洋张兴罗越王亚东赵彩彦
胡洋洋, 张兴, 罗越, 王亚东, 赵彩彦
河北医科大学第三医院湘江院区感染肝病科, 石家庄 050051
肝衰竭是由多种因素引起的肝脏严重损害,具有病情进展迅速、病死率高等特点。以免疫炎症损伤为核心的“二次打击学说”是肝衰竭发病关键机制。细胞自噬在肝脏免疫炎症损伤过程中发挥关键调控作用。近年研究表明自噬通过调节炎症小体活化、对抗氧化应激、抑制细胞凋亡等机制对肝衰竭发挥保护效应。本文就自噬在肝衰竭中的变化、调控机制及其作为肝衰竭治疗靶点的潜在前景作一综述。
1 自噬在肝衰竭中的变化特点
自噬是一种进化上高度保守的细胞适应性保护机制,由30余种自噬相关基因(autophagy related gene,Atg)及其编码的Atg蛋白分子家族所介导,参与细胞质质量控制、新陈代谢、免疫调控等过程。尤其选择性自噬通过降解受损细胞器,发挥细胞“清道夫”作用[1]。在肝衰竭的发生发展过程中存在自噬受损现象,而代偿性激活自噬可通过抑制肝细胞内蛋白质聚集、脂质堆积、氧化应激、细胞炎症与死亡,维持肝细胞生存稳态。
Ren等[2]研究证实,HBV相关急性肝衰竭(acute liver failure,ALF)患者肝组织自噬相关分子Atg7、Atg5、Beclin-1表达显著降低。同样,慢加急性肝衰竭(acute-on-chronic liver failure,ACLF)患者肝组织Atg5、Beclin-1、微管相关蛋白1轻链3(microtubule associated protein 1 light chain 3,LC3)-Ⅱ/LC3-Ⅰ的表达也显著降低,且伴随自噬底物连接蛋白p62聚集性升高[3]。Wu等[4]研究则发现终末期肝病模型(model for end-stage liver disease,MELD)评分>25的ACLF患者脂多糖(lipopolysaccharide,LPS)水平显著高于MELD评分≤25患者,进一步体外实验显示LPS通过p38MAPK信号通路抑制肝星状细胞(hepatic stellate cell,HSC)自噬,并伴随HSC来源的IL-1β及其前体表达增加。上述研究证实肝衰竭患者存在自噬受损并可因此诱发或加重肝脏炎症反应。由D-氨基半乳糖(D-galactosamine,D-GalN)/LPS或CCl4诱导的肝衰竭动物模型显示,在急性肝损伤(acute liver injury,ALI)早中期,肝脏自噬水平升高,当进展至肝衰竭阶段自噬水平却降低,激活自噬显著减轻肝脏炎症损伤,提高ALF小鼠存活率[2,5]。因此,自噬在肝衰竭发生发展不同阶段存在动态变化,且对肝衰竭发挥保护作用。在对乙酰氨基酚(acetaminophen,APAP)诱导的动物和肝细胞损伤模型中也均可观察到自噬激活现象,后者通过去除受损线粒体发挥肝脏保护作用[6-7]。但过量APAP通过损害自噬通量,导致受损线粒体不能被有效清除,增加线粒体氧化应激,造成肝细胞坏死[8]。值得注意的是,刀豆素A诱导的ALI小鼠模型中,自噬呈过度激活并诱导树突状细胞活化,加剧肝脏免疫炎症反应,甚至触发肝细胞自噬性死亡,小鼠存活率显著降低[9]。由此可见,虽然不同病因、不同阶段的肝衰竭存在自噬强度和方向的差异,也影响着肝衰竭疾病转归和结局,但更多证据支持适度自噬有利于限制肝衰竭发生发展。
2 自噬参与肝衰竭保护作用的机制
2.1 调节Nod样受体热蛋白结构域相关蛋白3(Nod-like receptor pyrin domain-containing protein 3,NLRP3)炎症小体信号 NLRP3炎症小体由Nod样受体、凋亡相关斑点样蛋白(apoptosis-associated speck-like protein containing a CARD,ASC)和半胱氨酸天冬氨酸蛋白酶-1(cysteiny aspartate-specific protease-1,caspase-1)组成,通过介导IL-1β和IL-18成熟信号,调节炎症和免疫反应。Jia等[10]发现HBV相关ACLF患者肝组织中NLRP3及caspase-1、IL-18和IL-1β表达水平升高,是造成肝脏持续性炎症损伤的重要原因。ALF小鼠模型肝组织中NLRP3、ASC、caspase-1等也显著升高,并伴随肝细胞空泡变性和坏死增加[11],因此NLRP3炎性小体激活是肝衰竭炎症损伤核心机制之一。
在D-GalN/LPS诱导的ALF小鼠模型中,应用组蛋白去乙酰化酶抑制剂CAY10683修饰自噬相关蛋白ULK1 K68赖氨酸位点,上调ULK1表达,继而抑制NLRP3活化改善肝功能[12]。应用LPS处理人HSC细胞株LX2后,Atg13、LC3-Ⅱ表达呈剂量依赖性下降,并伴随NLRP3、IL-1β及其前体的表达增加,当LX2细胞暴露于自噬抑制剂巴佛洛霉素A1可进一步促进p62积累、NLRP3活化和IL-1β产生[4]。上述研究均提示自噬参与对NLRP3炎性小体的调控,并因此影响肝脏免疫炎症损伤。此外,NF-κB参与上调NLRP3和IL-1β前体等转录表达,是NLRP3炎症小体激活的重要信号。Shan等[13]发现APAP诱导的ALI小鼠肝组织NLRP3、caspase-1、IL-1β表达均明显增加,自噬激活剂雷帕霉素(rapamycin,RAPA)可显著抑制APAP诱导的NF-κB核移位以及NLRP3通路活化。反之,抑制自噬则使IκBα受抑和p-NF-κBp65蛋白表达增加,激活NF-κB,上调NLRP3信号通路相关炎症因子的转录和翻译。
体外细胞研究[14]也证实,生长停滞特异蛋白6信号通路介导的自噬通过抑制肝脏Kupffer细胞中caspase-1的激活,抑制促炎因子生成,阻止肝脏炎症反应进展。此外,应用LPS刺激体外培养的单核细胞显示ASC呈p62依赖性降解,抑制炎症小体活性[15]。综上,自噬通过抑制NF-κB核移位减少炎症小体转录、抑制caspase-1活性以及吞噬和降解NLRP3炎性小体成分等机制负性调控NLRP3炎症小体活性,从而减轻肝衰竭炎症损伤。
2.2 抑制氧化应激 氧化应激介导的肝损伤也是肝衰竭发病机制的重要环节,由于机体氧化与抗氧化系统失衡,活性氧(reactive oxygen species,ROS)过度生成和积聚,从而造成肝细胞广泛而不可逆性死亡。在LPS/D-GalN诱导的ALF小鼠肝组织以及LPS处理的巨噬细胞RAW264.7和HSC中均可检测到大量ROS产生,从而消耗内源性抗氧化物质,导致核酸、蛋白质、脂质等细胞成分受损,加剧肝细胞死亡[16-17]。
研究发现,线粒体自噬通过选择性清除受损线粒体,减少APAP诱导的受损肝细胞ROS产生,减轻肝脏氧化应激性损伤。在自噬相关分子Atg5缺陷的败血症小鼠模型中,肝细胞内受损线粒体大量积累,通过增加线粒体ROS的产生并启动线粒体凋亡途径,加速器官功能衰竭[18]。自噬选择性清除功能障碍线粒体的机制复杂,可能与PTEN诱导性激酶蛋白1(PTEN induced putative kinase 1,PINK1)-Parkin信号通路相关。Wang等[19]通过电镜观察发现PINK1和Parkin双敲除小鼠在APAP处理后自噬小体和溶酶体数量减少,并且线粒体蛋白泛素化和p62蛋白移位明显减少,延迟肝脏谷胱甘肽还原,进而加速肝细胞坏死及小鼠死亡。自噬调节氧化应激的另一机制由核因子E2相关因子2(nuclear factor erythroid 2-related factor,Nrf2)介导。生理条件下,Nrf2与Kelch样ECH相关蛋白1结合而处于失活状态,当发生氧化应激时可被磷酸化的p62激活,继而结合细胞核抗氧化反应元件,上调抗氧化酶基因转录,参与细胞对氧化应激性损伤的防御。在过量APAP诱导的ALI早期,p62、磷酸化p62水平显著上调,并促进Nrf2向细胞核内移位,上调抗氧化基因血红素加氧酶-1、谷氨酸-半胱氨酸连接酶催化亚基(glutamate-cysteine ligase catalytic subunit,GCLC)等细胞保护酶表达,代偿性对抗氧化应激,下调肝细胞ROS水平,降低肝细胞损伤风险[20]。Ruart等[21]检测CCl4处理的大鼠肝窦内皮细胞显示早期阶段(4周)可观察到自噬增强,而晚期(6周)自噬强度不再变化。进一步通过敲除肝窦内皮细胞Atg7的动物模型证实,仅在肝窦内皮细胞中Nrf2依赖性抗氧化基因GCLC、谷胱甘肽-S-转移酶Mu等表达上调,提示自噬缺陷是Nrf2激活的重要触发因素。综上,在肝衰竭发病机制中自噬与Nrf2信号通路均参与调控氧化应激与抗氧化机制的平衡,并且在其调控过程中呈现时间和空间差异。
自噬对抗氧化应激的过程同时也参与NLRP3炎症小体活性调控,说明两种机制存在交叉。高浓度ROS可使硫氧还蛋白相互作用蛋白与硫氧还蛋白解离,直接与NLRP3相互作用,激活NLRP3炎症小体。Yang等[22]检测RAPA处理的肝损伤小鼠模型肝组织显示,增强的自噬能够清除过量释放的ROS,下调硫氧还蛋白相互作用蛋白/NLRP3轴,从而抑制小鼠肝脏中NLRP3炎症小体活化。由PINK1介导的线粒体自噬也可通过清除线粒体ROS抑制肝组织NLRP3炎症小体活化、线粒体氧化应激,从而减轻黄曲霉素B1诱导的小鼠肝组织炎症损伤[23]。上述研究揭示了自噬和氧化应激、NLRP3炎症小体之间的新联系,自噬在肝衰竭发病机制的调控中具有枢纽作用。
2.3 调控细胞凋亡 TNF/TNF受体系统激活介导的细胞凋亡同样是ALF发病重要分子机制之一。自噬调控细胞凋亡对维持细胞生存具有重要意义。Ahmedy等[24]在LPS/D-GalN诱导的ALF小鼠肝组织中观察到细胞凋亡增加,p53和caspase-3、Bax表达显著上调,而在诱导ALF发生前应用柚皮素可使升高的caspase-3、p53和Bax水平分别降低49%、34%和54%,并且这种抑制细胞凋亡的作用可被自噬抑制剂3-甲基腺嘌呤消除。不仅如此,自噬还可在转录水平抑制TNF-α等炎症因子表达,或抑制TNF-α介导的半胱氨酸蛋白酶激活途径保护肝细胞免于凋亡。另一方面,线粒体膜电位下降是细胞凋亡的早期事件,自噬抑制剂3-甲基腺嘌呤通过降低ALF体外模型细胞L-02线粒体膜电位,增加细胞凋亡相关蛋白Bax、细胞色素C水平,对抗自噬介导的抗肝细胞凋亡作用[25]。Oami等[18]通过透射电镜观察发现,条件性Atg5基因敲除小鼠的肝细胞线粒体损伤和细胞凋亡加速,可能由于自噬缺陷诱导了线粒体死亡途径,促进肝细胞凋亡。自噬不仅参与调控肝细胞凋亡,Tian等[26]发现抑制自噬也可以促进HSC凋亡,导致细胞外基质生成减少,加速ALF肝脏实质结构的塌陷;利用RAPA激活自噬可抑制一氧化氮介导的细胞凋亡,保护HSC,为再生肝细胞提供支架。以上研究表明,自噬参与肝细胞和HSC凋亡的调控过程以减轻肝组织损伤,并且TNF-α、线粒体膜电位、一氧化氮等分子信号在其中发挥重要作用(图1)。
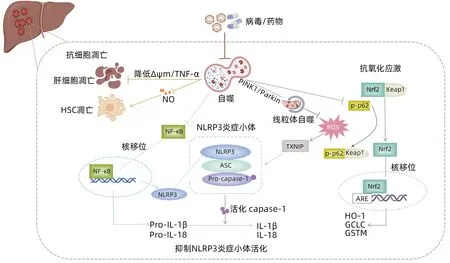
图1 自噬在肝衰竭中的保护作用机制Figure 1 The protective mechanism of autophagy in liver failure
3 调控自噬对肝衰竭治疗的价值
3.1 外泌体与自噬 外泌体是膜衍生的纳米级囊泡(直径为30~150 nm),可携带蛋白质、脂类和核酸等多种组分,参与清除致病或受损的细胞内物质、介导细胞间通讯[27-28]。越来越多研究[29]证实,自噬与外泌体存在交互作用,自噬可以降解功能失调的外泌体,而外泌体又可以通过诱导细胞自噬发挥细胞保护作用。Shen等[30]建立Atg5-/-AML12细胞模型显示,经IL-1β/TNF处理后的AML12细胞可分离出含损伤相关分子模式的外泌体,后者进一步上调Kupffer细胞IL-1、IL-6等基因表达,导致细胞能量稳态失衡、溶酶体通透改变,加重炎症反应。国内学者最新临床研究[31-33]证实肝衰竭患者肝脏来源的外泌体数量、组分及功能方面均存在差异性变化,且影响着肝衰竭患者疾病严重程度和预后转归。
Yang等[34]体外诱导骨髓间充质干细胞分化为肝细胞样细胞后分离提取外泌体,并将其应用于肝脏缺血再灌注小鼠模型显示,处理后小鼠肝组织LC3-Ⅱ、Beclin-1水平升高,p62/SQSTM1水平降低,抑制肝细胞变性和坏死,证实外泌体可以通过增强自噬减轻肝脏损伤。此外,人骨髓间充质干细胞来源外泌体通过输送微小核糖核酸let-7a-5p靶向MAP4K3-转录因子EB信号通路恢复ACLF小鼠肝组织自噬通量,有效减轻肝细胞损伤和凋亡[35]。综上,自噬缺陷可能促进肝细胞分泌损伤相关分子模式外泌体,激活巨噬细胞引起炎症,而干细胞来源的外泌体则通过增强自噬发挥肝脏保护作用,为临床应用外泌体治疗肝衰竭提供策略。
3.2 过氧化物酶体增殖物激活受体α(PPARα)与自噬 PPARα激动剂亦可通过促进ALF小鼠Atg5、Atg7、溶酶体膜相关蛋白-1和LC3-Ⅱ表达,延缓ALF进展;抑制自噬则逆转PPARα的肝脏保护作用,促进炎症因子和趋化因子表达上调,加剧肝脏炎症损伤。Ren等[2]证实抑制糖原合成酶3β(glycogen synthase kinase 3β,GSK3β)活性可通过增加PPARα表达激活自噬减轻肝脏损伤,应用siRNA沉默PPARα表达则逆转上述作用。磷酸腺苷活化蛋白激酶(AMP-activated protein kinase,AMPK)也通过影响PPARα活性参与肝衰竭自噬激活。Liu等[36]报道在miR-19a调控的肝细胞AMPK/PPARα自噬信号通路中,miR-19a通过激活AMPK增强PPARα的转录活性上调LC3-Ⅱ和Beclin-1表达。此外,PPARα激动剂非诺贝特通过激活自噬、降低肝脏ROS和脂质过氧化有效减轻APAP诱导的ALI。因此,调控PPARα活性为靶点的信号分子可为激活自噬发挥肝衰竭保护作用提供新思路。
3.3 其他 自噬作为拮抗肝细胞炎症和坏死的核心机制,还受其他多种分子信号调控。例如,RAPA靶蛋白抑制剂西罗莫司可增强自噬,减轻肝细胞损伤与死亡,目前已在动物或体外实验模型中应用[22,37]。烟酰胺磷酸核糖转移酶抑制剂FK866通过抑制c-Jun氨基末端激酶信号通路诱导自噬,对D-GalN/LPS和刀豆素A诱导的ALF小鼠发挥保护作用[38]。可溶性T细胞免疫球蛋白黏蛋白分子3也可以增加ALF小鼠肝脏CD11b+巨噬细胞中LC3-Ⅱ的水平,促进自噬小体形成,减少肝脏炎症介质产生[39]。骨髓间充质干细胞也可以通过PI3K/Akt途径调节自噬以减轻ALF肝细胞凋亡与肝组织炎症反应,而其旁分泌的血红素加氧酶-1也参与自噬的调节[40]。虽然上述研究尚有待进一步深入,但相关机制探索为临床发掘基于自噬调控的分子靶向治疗提供了一系列的研究思路和开发靶位。
4 结语与展望
综上,自噬是保护肝细胞免受炎症损伤的重要机制。靶向激活自噬将可能成为肝衰竭治疗领域的重要课题。虽然目前将调控自噬作为肝衰竭治疗的靶点尚存在诸多问题和体内实践应用的风险,包括:(1)目前研究大多局限于细胞和动物模型,在肝衰竭特定的炎症背景下激活自噬的具体分子调控机制和信号通路仍待深入;(2)自噬信号通路涉及多种分子调节机制,但目前尚未评价出最佳分子调控靶位;(3)尚无特异性靶向肝脏的自噬激活剂,在激活肝脏自噬的同时可能使患者面临其他脏器细胞自噬性死亡和肿瘤发生的风险;(4)自噬具有抑制HSC凋亡为再生细胞提供支架作用,但HSC凋亡过度抑制可能导致肝纤维化加重,降低自噬激活剂在临床应用的安全性。但自噬通过参与调控NLRP3炎症小体介导的炎症反应、对抗氧化应激、抑制细胞凋亡等方式发挥对肝衰竭的保护作用毋庸置疑,以调控外泌体、PPARα等为靶点的自噬活化策略也将有望成为肝衰竭分子靶向治疗的重要策略。
利益冲突声明:本文不存在任何利益冲突。
作者贡献声明:胡洋洋负责查阅文献,撰写文章;张兴、罗越负责校对文章;王亚东负责指导立题及修改;赵彩彦负责审阅文章等。
