Effect of aggregation on thermally activated delayed fluorescence and ultralong organic phosphorescence: QM/MM study
2023-11-02QunZhang张群XiaofeiWang王晓菲ZhiminWu吴智敏XiaofangLi李小芳KaiZhang张凯YuzhiSong宋玉志JianzhongFan范建忠ChuanKuiWang王传奎andLiliLin蔺丽丽
Qun Zhang(张群), Xiaofei Wang(王晓菲), Zhimin Wu(吴智敏), Xiaofang Li(李小芳), Kai Zhang(张凯),Yuzhi Song(宋玉志), Jianzhong Fan(范建忠), Chuan-Kui Wang(王传奎), and Lili Lin(蔺丽丽)
Shandong Key Laboratory of Medical Physics and Image Processing&Shandong Provincial Engineering and Technical Center of Light Manipulations,School of Physics and Electronics,Shandong Normal University,Jinan 250358,China
Keywords: organic light-emitting diodes, thermally activated delayed fluorescence, ultralong organic phosphorescence,aggregation mode
1.Introduction
In the past few years, extensive attention has been paid to organic thermally activated delayed fluorescence (TADF)emitters due to their unique photophysical properties.[1-4]Since Pope and others first reported organic electroluminescent materials in 1963,[5]organic electroluminescent materials have gradually entered the public’s field of vision, especially in the fields of solid-state lighting, display panels,organic light-emitting diode (OLED) TV, digital cameras,and so on.[6-8]TADF materials are generally supposed to be the third generation of organic light-emitting materials after traditional fluorescent materials and traditional phosphorescent materials, with their internal quantum efficiency (IQE)reaching 100%.[9-16]For TADF materials,the excitons in the triplet state cross to the singlet state through reverse intersystem crossing (RISC) on thermal activation, and thereafter emit delayed fluorescence from the singlet state to the ground state.The implementation of the TADF process requires a small energy gap between the first singlet excited state and the first triplet excited state (ΔEst), which can be realized by a small overlap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO).The general method to obtain TADF is to use a molecular design strategy combining donor and acceptor groups.[17-22]Of course, molecular aggregation behavior can also induce and regulate the TADF process.In recent years,there have been preliminary studies on the law of accumulation modes in TADF behavior.It is found that J-aggregation can reduce ΔEst,which promotes the RISC process and TADF process, in general.H-aggregation promotes the intersystem crossing(ISC)process and weakens TADF,but promotes the RTP process.[23]In 2019, Thilagaret al.reported that a molecule crystallized in two different crystal forms (1a and 1b).[24]Polymorph 1b with J-aggregation has brighter TADF phenomenon than polymorph 1a with a random arrangement.In the same year, Yanget al.reported two DMAC-CNQ and FDMAC-CNQ molecules with multi-functional emission,and realized TADF,AIE and MCL by regulating their aggregation mode.[25]In 2021, Yanget al.found the regulation of aggregation state on TADF in TRZ-c-BPXZ and TRZ-a-BPXZ molecules.[26]In the same year,Fuet al.found J-aggregation state regulation on TADF in the (E)-5-(ethoxycarbonyl)-2,2-difluoro-6-methyl-4-(2-[1-methyl-1H-indol-2-yl]vinyl)-2H-1,3,2-dioxaborinin-1-ium-2-uide molecule.[27]However,there are few studies of the aggregation effect on TADF properties theoretically, due to the difficulty in the simulation of aggregation states.In addition, the complicity in aggregation states makes it more difficult to understand the light-emitting properties.
Recently, a pair of chiral TADF molecules(R)-5-(9H-carbazol-9-yl)-2-(1,2,3,4-tetrahydronaphthalen-1-yl)isoindoline-1,3-dione [(R)-lmNCz] and (S)-5-(9H-carbazol-9-yl)-2-(1,2,3,4-tetrahydronaphthalen-1-yl)isoindoline-1,3-dione[(S)-lmNCz](as shown in Figs.1(a)-(d)) have been reported.[28]The block-like single crystals(SCb-form) and prism-like single crystals (SCp-form) of(R/S)-ImNCz are found to have different luminescent properties.The multiple ultralong phosphorescence (UOP) peaks in the spectrum were of complex origins,and they are related not only to ImNCz but also to a small amount of impurities(ImNBd) in the crystal prepared in the laboratory.There is no TADF phenomenon in the SCb-form, while TADF phenomenon is found in the SCp-form.We believe that this is related to their different aggregation methods.The SCb-form shows face-to-face stacking and the SCp-form adopts edge stacking.The SCpcrystal has a wide frequency band in the range of 430-520 nm and three peaks in the range of 520-700 nm.In order to verify the generation mechanism of TADF and RTP in SCpcrystals and the absence of TADF in SCbcrystals, the luminescence characteristics of the chiral molecules in the crystalline state are studied by combining quantum mechanics and molecular mechanics(QM/MM)methods.At the same time,the luminescence characteristics of(R)-lmNCz,(S)-lmNCz,and(R)-lmNBd in acetonitrile solution are studied using the polarizable continuum model (PCM).Recently, Li and Zhanget al.performed theoretical studies on SOBF-OMe and DBPz-2spAc with PCM and QM/MM methods,and good agreement with experimental results was obtained, which indicates the reliability of this method.[29,30]Nevertheless,these calculation models were limited to only the ONIOM model of single components.In this study, the impurity in the crystal is also considered to explain the emission spectra,which indicates that the impurity could also participate in the emission.The photoluminescence properties of(R)-lmNCz,(S)-lmNCz,and(R)-lmNBd in gas phase are also studied.Both monomers and dimers are selected as research models to explore the origin of TADF.Our existing calculation results demonstrate that the generation of broadband is closely related to the radiation transition of S1.The three peaks in the range of 520-700 nm(as shown in Fig.1(e)) should be due to a small amount of(R)-lmNBd impurities in the crystals prepared in the laboratory,which agree with experimental guess.[31]Our theoretical simulation results provide a reasonable explanation for the generation mechanism of TADF and RTP, which is helpful to understand the influence of molecular stacking mode on luminescence properties.

Fig.1.Molecular structures and ONIOM model of(R)-lmNCz-SCb,(S)-lmNCz-SCb,(R)-lmNCz-SCp,(S)-lmNCz-SCp,and(R)-lmNBd.The centered molecule is treated as the high layer and surrounding molecules are regarded as the low layer.
2.Theoretical methods and calculation details
In this work, the luminescence characteristics of (R)-lmNCz, (S)-lmNCz, and (R)-lmNBd in acetonitrile solution are simulated with the PCM method which has turned out to be a trustworthy tool for predicting the influence of solvents on TADF molecules.[32,33]The calculation model is established according to the four crystal structures obtained from the experiment(as shown in Fig.1).[28]A two-layer ONIOM method is adopted for the molecular crystal simulation.[34-37]The central molecule is chosen as the high layer and measured by the QM method, while the surrounding molecules are selected as the low layer and determined by the MM method.The calculation of the ground state and excited state of the high layer molecule in the center is based on density functional theory (DFT) and time-dependent density functional theory(TD-DFT), respectively.[38]For the surrounding molecules,the universal force field(UFF)which sustains major elements is applied, which has also been proven reliable.[39,40]In the optimization process, the central molecule is completely released, while other molecules are locked.In our study, the ONIOM model of(R)-lmNBd(as shown in Fig.1(e))is constructed by replacing a(R)-lmNCz molecule in the SCpcrystal of (R)-lmNCz, and the replacement (R)-lmNBd is set as the central molecule in the ONIOM model.For the sake of researching the interaction between (R)-lmNCz molecules and surrounding(R)-lmNCz molecules,a dimer is taken as central molecule based on the ONIOM model.In order to study the interaction between the (R)-lmNBd molecule and surrounding (R)-lmNCz molecules, an (R)-lmNBd molecule and an(R)-lmNCz molecule are taken as a complex in the ONIOM model.The geometries of the dimer and complex are shown in Fig.S1.The intermolecular interactions of (R)-lmNCz,(S)-lmNCz, and (R)-lmNBd are assessed by the independent gradient model based on Hirshfeld partition(IGMH).[41]QM/MM methods are also used to calculate the photophysical properties of the dimer and the complex.Five functionals(WB97XD,PBE0, M062X,BMK,and B3LYP)are tested on the emission wavelengths together with a 6-31G*basis set(as shown in Table 1).[42-46]Functional fitting results show that the fluorescence wavelengths of(R)-lmNCz-SCb,(S)-lmNCz-SCb, (R)-lmNCz-SCp, and (S)-lmNCz-SCpfit well with the experimental values at the PBE0/6-31G*level.Therefore,all the operations of the following QM parts are performed at the PBE0/6-31 G*level.All these corresponding calculations are implemented in the Gaussian 16 package.[47]

Table 1.Emission wavelengths calculated by different functionals for (R)-lmNCz-SCb, (S)-lmNCz-SCb, (R)-lmNCz-SCp,and(S)-lmNCz-SCp in the solid phase(unit: nm).
Efficient intersystem crossing rates and reverse intersystem crossing rates (KISC,KRISC) are significant indicators to measure the TADF process and the RTP process.TheKISCbetween two electronic states with different spin states can be described as[48,49]
whereZiis the partition function andρISC(t,T)is the thermal vibration correlation function.〈Φf| ˆHSO|Φi〉is the spin-orbit coupling(SOC)between two states with different spin multiplicity,which can be calculated by quadratic response function methods in the Dalton Program.[50]KRISCcan also be calculated making use of Eq.(1).The ISC and RISC rates are all calculated using molecular materials property prediction package(MOMAP).[51]
In addition,the radiation rate(Kr)is calculated by the Einstein spontaneous emission equation[52]
wherefis oscillator strength and ΔEfiis the vertical emission energy between the first singlet excited state(S1)and ground state(S0)(or between the first triplet excited state(T1)and S0)in the units of wavenumber(cm-1).
Furthermore, the non-radiative decay rate from S1to S0(K1nr)can be deduced based on first-order perturbation theory and Fermi’s golden rule(FGR).Through Fourier transform,it can be written as

As for the non-radiative decay rate from T1to S0(),it can be calculated by the following formula:[54]
Here, ΔEadrepresents the adiabatic excitation energy andλjrefers to the reorganization energy of thejth vibrational mode.|〈S0| ˆHSO|Tn〉|is the SOC matrix element between S0and Tn.
The Huang-Rhys factor and recombination energy are obtained using the DUSHIN software package for normal mode analyses on the obtained geometric structure and electronic structure information.The Huang-Rhys factor and reorganization energy formula can be written as
Hereωkrefers to the vibration frequency andDkrepresents the normal coordinate displacement of modek.
In addition, for identification and visualization of the weak interaction between two molecules in an aggregate state,a function(δg)is used to define the intermolecular interaction based on IGMH:
Here,AandBrefer to the two fragments in which the interaction is to be investigated,andiandjrepresent the atoms in these two fragments,respectively.The atomic pairδgindex is denoted asδGpair.Accordingly,δGatom(%)is defined to survey the percentage of an atom’s contribution to the interaction between fragments:
3.Results and discussion
3.1.Molecular structures
The molecular structures of(R)-lmNCz-SCb,(S)-lmNCz-SCb, (R)-lmNCz-SCp, (S)-lmNCz-SCp, and (R)-lmNBd are shown in Fig.1.The molecular structures of complex and dimer are shown in Fig.S1.The geometry of the ground state (S0), the first singlet excited state (S1), the first triplet excited state (T1), and the second triplet excited state (T2)of (R)-lmNCz, (S)-lmNCz, and (R)-lmNBd molecules in the gas phase,acetonitrile solution,and solid phase are optimized.The calculated properties of the(R)-lmNCz molecule and the(S)-lmNCz molecule in the gas phase and in solution are very similar(see Table S1).The properties of(R)-lmNCz and(S)-lmNCz in the same crystal structure are also very similar, so(R)-lmNCz-SCband(R)-lmNCz-SCpare selected for the next analysis.The relevant calculation results of (S)-lmNCz-SCband(S)-lmNCz-SCpare shown in Figs.S2-S9 and Tables S1-S6.
The root of the mean squared displacement(RMSD)can be used to describe the configuration difference between the ground state and the excited state for (R)-lmNCz-SCb, (R)-lmNCz-SCp,and(R)-lmNBd,as shown in Fig.2.Tables S7-S11 list some important geometric parameters and experimental data of(R)-lmNCz-SCb,(R)-lmNCz-SCp,(R)-lmNBd,complex,and dimer molecules in the S0,S1,T1,and T2states.As shown in Fig.2, the configuration changes between S0and S1show that distinct aggregation methods and distinct donors have different effects on the geometric changes during molecular excitation, where the RMSD values of (R)-lmNCz-SCb, (R)-lmNCz-SCp, and (R)-lmNBd are 0.082 °A,0.077 °A, and 0.135 °A, respectively.In different aggregation modes, the bond lengths and bond angles of (R)-lmNCz-SCband(R)-lmNCz-SCpare similar,but the dihedral angles of(R)-lmNCz-SCband(R)-lmNCz-SCpare significantly different(as shown in Table S7 and S8).The change in dihedral angles of(R)-lmNCz-SCb(α1changes from-6.0°to 4.4°,α2changes from 2.1°to-0.8°) is significantly greater than that of (R)-lmNCz-SCp(α1changes from-1.7°to 1.9°,α2changes from-3.9°to-0.2°)when they are changed from S0to S1.Therefore,the larger dihedral angle change of(R)-lmNCz-SCbmay increase the reorganization energy of a molecule.For (R)-lmNBd, the change in bond lengths and bond angles is also very small, mainly locating at the change of dihedral angles(α1changes from 16.3°to 8.8°,α2changes from-21.7°to-8.9°), which may make the non-radiative rate between S0and S1state of(R)-lmNBd very large.In contrast,the dihedral angles of (R)-lmNCz-SCb(α1changes from 4.4°to 1.1°,α2changes from-0.8°to-4.5°), (R)-lmNCz-SCp(α1changes from-1.7°to 1.9°,α2changes from-0.8°to-4.5°), and(R)-lmNBd (α1changes from 8.8°to 7.1°,α2changes from-8.9°to-12.5°) have small differences, no more than 10°,in the S1and T1state, which means their RMSD values are also smaller.The geometric difference between (R)-lmNCz-SCband(R)-lmNCz-SCpin the S1and the T2state are small,and their RMSD values are also very small.This predicts a possible transition path from the high-energy excited state T2to the S1state.Dihedral angles of (R)-lmNBd,α1andα2,change greatly in the S1and the T2state withα1changed by nearly 5.5°andα2changed by nearly 10.7°, so the RMSD values of the S1and the T2state of (R)-lmNBd are large.At the same time, by analyzing the important geometric parameters of complex and dimer, it is found that the bond length and bond angle before and after optimization are little different,but the dihedral angle has changed.Therefore,we believe that the different aggregation modes and the changes of donor group will have a certain impact on the interaction between molecules,and then affect the molecular configuration.
Figure 3 shows the energy and distribution of frontier molecular orbitals.It is easy to find that the HOMO of (R)-lmNCz is mainly concentrated in the carbazole group, and the LUMO is mainly located in the phthalimide group.The HOMO-LUMO overlap of (R)-lmNCz-SCpis smaller than that of (R)-lmNCz-SCb, which is related to the fact that the dihedral angle(-61.94°)between the phthalimide group and carbazole group of (R)-lmNCz-SCpis larger than the dihedral angle(-46.99°)between the phthalimide group and carbazole group of (R)-lmNCz-SCb.This may be due to a stronger intermolecular interaction in the SCpcrystal, which makes the conformation of (R)-lmNCz-SCpmolecule more distorted.The large dihedral angle makes the molecular conformation more distorted and the HOMO-LUMO overlap is small, which is conducive to reducing ΔEst.The HOMO of(R)-lmNBd is mainly distributed in the benzoindole group,and the LUMO is mainly concentrated in the phthalimide group.Obviously, the dihedral angle between the phthalimide group and the benzoindole group in the(R)-lmNBd molecule is large(-79.77°),thus the HOMO-LUMO overlap is small and ΔEstis also small.The HOMO-LUMO energy gap of(R)-lmNBd(2.393 eV) is obviously smaller than that of (R)-lmNCz-SCb(3.072 eV) and (R)-lmNCz-SCp(2.871 eV), so the emission of (R)-lmNBd has red shift.The frontier molecular orbital analysis of complex and dimer shows that the HOMO-LUMO distributions of the complex are analogous to those of (R)-lmNBd,and the HOMO-LUMO distributions of the dimer are similar to those of (R)-lmNCz-SCp.It is found that the (R)-lmNBd will induce a smaller HOMO-LUMO energy gap.
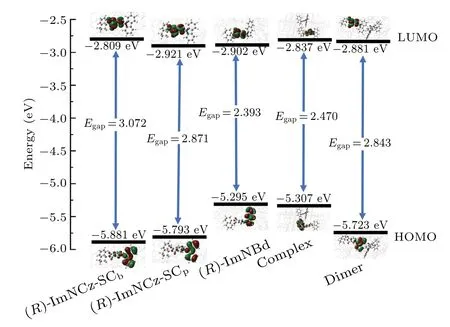
Fig.3.Energies and electronic distributions of HOMO and LUMO for(R)-lmNCz-SCb, (R)-lmNCz-SCp, (R)-lmNBd, complex, and dimer in S1 state in the solid phase.
3.2.Excited states
It is well known that the small energy gap between S1and Tnin TADF molecules and RTP molecules is an important parameter for rapid RISC and ISC processes.The adiabatic excitation energies of(R)-lmNCz in the two aggregation modes and (R)-lmNBd, complex, and dimer in the ONIOM model are shown in Table 2 and Fig.4.The results reflect that the energy gaps between S1and S0of (R)-lmNCz-SCband(R)-lmNCz-SCp(2.49 eV and 2.50 eV)are roughly equal,but the ΔEstof (R)-lmNCz-SCp(0.13 eV) is smaller than that of(R)-lmNCz-SCb(0.21 eV) due to the higher triplet energy in SCpmode, which makes (R)-lmNCz-SCpmore likely to exhibit TADF than (R)-lmNCz-SCb.For (R)-lmNBd, both S1and T1become lower than (R)-lmNCz.The energy gap between them is also smaller.The energy gap between T1and S0of(R)-lmNBd(1.97 eV)is small,thus(R)-lmNBd is more likely to emit phosphorescence.Although the energy gap between S1and S0of the dimer (2.38 eV) is larger than that between S1and S0of the complex (2.25 eV), the ΔEstof the dimer(0.13 eV)is smaller than that of the complex(0.16 eV),which makes the dimer more likely to display TADF than the complex.It is found that the energy gaps between T1and S0of the(R)-lmNBd,complex,and dimer increase in turn(1.97 eV,2.09 eV,and 2.25 eV),indicating that the emission wavelength would shift blue.Introducing(R)-lmNBd would induce lower excited states,and thus red shift emission.
Besides ΔEst,the transition properties and SOC constants between the two electronic states are also important indicators to determine RISC and ISC processes.By analyzing the S1, T1, and T2states of (R)-lmNCz-SCb, (R)-lmNCz-SCp,(R)-lmNBd, complex, and dimer, the natural transition orbit(NTO)shown in Fig.5 is obtained.All S1states and T2states have charge transfer (CT) characteristics, and T1states have hybridized local charge-transfer (HLCT) properties.The results show that there is only partial transfer between the acceptor group and the donor group of the T1state of (R)-lmNCz-SCband (R)-lmNCz-SCpmolecules, which would make the SOC between the S1state and the T1state larger.This helps to promote the intersystem crossover of excitons between the S1state and the T1state.The CT feature of the S1state is helpful to reduce the ΔEst.It is found that different stacking patterns have little influence on the transition properties of the triplet excited states.

Table 2.Calculated emission wavelength for fluorescence(λfl)and photoluminescence(λph),oscillator strength(f),transition dipole moment(µ), adiabatic singlet (ES1) and triplet (ET1) energies, adiabatic singlet-triplet energy gap (ΔEst) of (R)-lmNCz-SCb, (R)-lmNCz-SCp, (R)-lmNBd,complex,and dimer in the solid phase.
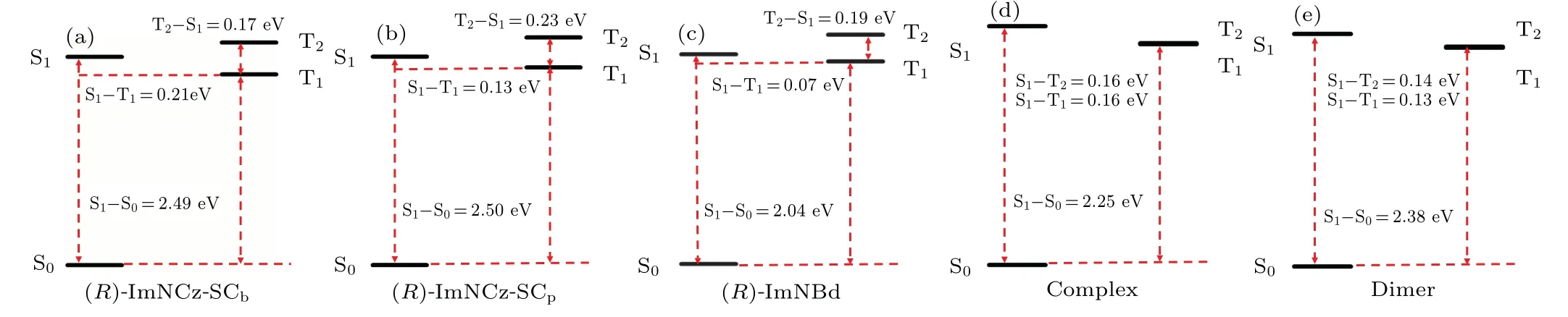
Fig.4.Excitation energy for(R)-lmNCz-SCb (a),(R)-lmNCz-SCp (b),(R)-lmNBd(c),complex(d),and dimer(e)in the solid phase.

Fig.5.Natural transition orbitals(NTOs)of the S1,T1,and T2 states based on their optimized structures for(R)-lmNCz-SCb (a),(R)-lmNCz-SCp (b),(R)-lmNBd(c),complex(d),and dimer(e)in the solid phase.
3.3.Reorganization energy and intermolecular interactions
The non-radiative decay process between S1and S0and between T1and S0are closely related to structural transformation and electron-vibration coupling.For the sake of further exploring the non-radiative process under diverse aggregation modes, the relationships between the frequency and the reorganization energy of(R)-lmNCz-SCb,(R)-lmNCz-SCp,and(R)-lmNBd in S1and T1are shown in Figs.6 and 7.The main dissipation channels of vibration relaxation can be divided into three parts: low-frequency(LF,<500 cm-1)mode,midfrequency (MF, 500-1500 cm-1) mode, and high-frequency(HF,>1500 cm-1)mode.The reorganization energy between S1and S0of (R)-lmNCz-SCpis mainly contributed by MF and HF modes.Compared with (R)-lmNCz-SCp, the reorganization energy between S1and S0of (R)-lmNCz-SCbin LF mode is increased, especially the rotational contribution between the dihedral angle of donor and acceptor, such as 161.58 cm-1.The increase in the dissipative pathway will increase the non-radiative decay process of molecules.In the(R)-lmNBd molecule, the reorganization energy between S1and S0is mainly contributed by LF and HF modes.In the LF mode,the bending vibration of the Bd group outward along the plane is the main contribution (115.91 cm-1), while the contribution in the HF mode mainly comes from C=O stretching vibration(1902.15 cm-1).The vibration modes of(R)-lmNBd between S1and S0and between T1and S0are similar,but the reorganization energy between T1and S0is significantly reduced,which is conducive to reducing the non-radiative dissipation from T1to S0.Moreover,the C-H stretching vibration at about 3500 cm-1also contributes to the reorganization energy for(R)-lmNBd.

Fig.6.Calculated reorganization energies versus the normal mode frequencies for(R)-lmNCz-SCb (a),(R)-lmNCz-SCp (b),and(R)-lmNBd(c)of the S1 state in the solid phase.Representative vibration modes are shown as insets.

Fig.7.Calculated reorganization energies versus the normal mode frequencies for(R)-lmNCz-SCb (a),(R)-lmNCz-SCp (b),and(R)-lmNBd(c)of the T1 state in the solid phase.Representative vibration modes are shown as insets.
In order to study the influence of intermolecular stacking mode on intermolecular interactions, we used IGMH analysis to visually analyze weak interactions(as shown in Fig.8).Although (R)-lmNCz-SCband (R)-lmNCz-SCpare the same molecule,they do not stack in the same way.It can be seen that in the SCbcrystal, the intermolecular interactions of dimer-4(-103.27 kJ/mol) and dimer-7 (-112.18 kJ/mol) are significantly stronger than those of other dimers, with obviousπ-πstacking and some C-H-πinteractions and C-H-O interactions.The large area in green between the adjacent phenyls of these two pairs of dimers shows that their intermolecular interactions are van der Waals interactions.In addition, it also shows representative characteristics ofπ-πstacking.The intermolecular interactions of dimer-6 (-3.64 kJ/mol) are the weakest.In other dimers, the intermolecular interactions are roughly the same.It is found that dimer-1 (-44.05 kJ/mol)and dimer-5(-45.36 kJ/mol)are mainly carbazole-carbazole interactions, and dimer-2 (-30.76 kJ/mol) and dimer-3(-31.32 kJ/mol) are mainlyπ-πstackings.In SCpcrystal, dimer-1 (-45.34 kJ/mol) has the strongest intermolecular interactions, including carbazole-carbazole interactions,C-H-πinteractions, and C-H-O interactions.It can be seen that dimer-5 (-38.24 kJ/mol) and dimer-7 (-38.25 kJ/mol)are mainly carbazole-carbazole interactions, and dimer-1(-36.53 kJ/mol) and dimer-3 (-6.53 kJ/mol) are mainly C-H-πinteractions.There are weak carbazole-carbazole interactions between dimer-4 (-13.78 kJ/mol) and dimer-6(-13.79 kJ/mol).We find that the strong intermolecular carbazole-carbazole interactions are conducive to the suppression of non-radiative channels.At the same time, we also analyzed the intermolecular interactions of the (R)-lmNBd molecule containing Bd impurity.The intermolecular interactions of dimer-1 (-47.99 kJ/mol) are the strongest, which are C-H-O interactions and C-H-πinteractions.Dimer-2 (-31.02 kJ/mol) has strong C-H-Bd interactions, dimer-3 (-22.12 kJ/mol) and dimer-4 (-19.23 kJ/mol) are similar to dimer-2, but C-H-Bd interactions are weak.There are many Cz-Bd interactions in dimer-5(-33.25 kJ/mol).dimer-6(-8.98 kJ/mol)and dimer-7(-8.12 kJ/mol)have weak Cz-Bd interactions.Obviously, the van der Waals interaction is the most important intermolecular interaction of the three systems.The face-to-face stacking mode in the SCbcrystal makes the system form intermolecular interactions with a strongππstacking effect.The edge stacking mode of the SCpcrystal makes the system form obvious intermolecular carbazolecarbazole interactions.The impurity system we simulated has a special intermolecular Cz-Bd interaction.

Fig.8.Intermolecular interactions for selected dimers of (R)-lmNCz-SCb (a), (R)-lmNCz-SCp (b), and (R)-lmNBd (c) described by the IGMH method.
3.4.Excited state dynamics
In order to quantitatively analyze the photophysical properties of molecules, we calculated the SOC constant at room temperature, as shown in Table 3.For (R)-lmNCz-SCp,the SOC constant between S1and T1(0.687 cm-1and 0.492 cm-1) is much larger than that SOC constant between S1and T2(0.069 cm-1and 0.108 cm-1),so the ISC processes and RISC processes occur more easily between S1and T1.(R)-lmNCz-SCbis similar to (R)-lmNCz-SCp, and the SOC constant between S1and T1(0.588 cm-1and 0.477 cm-1)of (R)-lmNCz-SCbis much larger than that between S1and T2(0.081 cm-1and 0.090 cm-1), so the ISC processes and RISC processes occur more easily between S1and T1.It can be seen that the SOC constant of S1-T1for (R)-lmNCz-SCpbased on the S1configuration(0.687 cm-1)is greater than that of (R)-lmNCz-SCb(0.588 cm-1), which is prone to ISC and RISC processes.Therefore,it is reasonable to believe that the fluorescence peak measured in the(R)-lmNCz-SCpcrystal experiment partly comes from the transition of excitons from the T1state to the S1state, then radiates to the ground state for luminescence.For(R)-lmNCz-SCpand(R)-lmNBd,the SOC constants between S0and T1calculated based on the S0configuration (2.782 cm-1and 1.458 cm-1) are also very large,which makes it possible to produce phosphorescence.
In addition,the calculated emission wavelength for fluorescence(λfl)and photoluminescence(λph),oscillator strength(f), transition dipole moment (µ) of (R)-lmNCz-SCb, (R)-lmNCz-SCp, (R)-lmNBd, complex, and dimer in the solid phase are shown in Table 2.It can be seen that the 430-520 nm fluorescence peak of the crystal in the experiment may be the joint action of(R)-lmNCz-SCp(496 nm)and dimer(521 nm).In the experiment,the three phosphorescence peaks of the crystal come from various sources.The phosphorescence emission peak at 550 nm may be the joint action of(R)-lmNCz-SCp(523 nm) and dimer (551 nm).The phosphorescence emission peak at 600 nm may be due to complex(592 nm)effect.The phosphorescence emission peak at 650 nm may be caused by (R)-lmNBd (630 nm).The phosphorescence peak at 550 nm has more sources than the ones at 600 nm and 650 nm, so the peak at 550 nm is more obvious.At the same time, we calculatedKr,Knr,KISC, andKRISC,as shown in Tables 4 and 5.TheK1rof(R)-lmNCz-SCp(6.94×107s-1) is obviously larger than that of (R)-lmNCz-SCb(1.12×107s-1), so it is easier for (R)-lmNCz-SCpto fluoresce.At the same time,K1ISCandK1RISCof (R)-lmNCz-SCp(1.83×108s-1,1.93×106s-1)are also very large,which is conducive to enhancing fluorescence emission.K2randK2nrof (R)-lmNCz-SCpand (R)-lmNBd are relatively small(2.6×100s-1, 1.04×103s-1s-1, 1.9×10-1, 4.95×105s-1),but the intersystem transition process from the S1state to the T1state of (R)-lmNCz-SCpand (R)-lmNBd (1.83×108s-1and 2.81×108s-1, respectively) are fast, which makes excitons accumulate in the T1state and promotes the T1-S0radiative process.

Table 3.Calculated SOC constants between S1 and T1,between S0 and T1,and between S1 and T2 for(R)-lmNCz-SCb,(R)-lmNCz-SCp,and(R)-lmNBd in the solid phase based on optimized S1,T1,and T2 structures.

Table 4.Rate constants of radiative and non-radiative between S0 and S1 (K1r,K1nr)and between S0 and T1 (K2r,K2nr)for(R)-lmNCz-SCb,(R)-lmNCz-SCp,and(R)-lmNBd in the solid phase.

Table 5.Effective intersystem crossing(ISC)rates and reverse intersystem crossing(RISC)rates between S1 and T1 (K1ISC,K1RISC)and between S1 and T2 (K2ISC, K2RISC) for (R)-lmNCz-SCb, (R)-lmNCz-SCp, and (R)-lmNBd in the solid phase.
4.Conclusions
In conclusion,the photophysical properties of(R)-lmNCz in two different aggregation modes are systematically studied based on QM/MM calculations, and the sources of multiple emission peaks detected in experiments are explained.The results show that the aggregation modes can lead to different energy diagrams of electronic states and emission wavelengths.The excited states of (R)-lmNCz-SCband (R)-lmNCz-SCphave similar transition properties, but (R)-lmNCz-SCbhas more vibrational modes than(R)-lmNCz-SCp,which increases the reorganization energy of (R)-lmNCz-SCband increases non-radiative process.For (R)-lmNCz-SCp, larger SOC and smaller ΔEstfavor the generation of TADF.In addition, it is verified that the generation of a phosphorescence peak in the SCpcrystal may be related to a small amount of (R)-lmNBd impurity in the crystal.The phosphorescence peak at 550 nm has one more source than that at 600 nm and 650 nm, so the phosphorescence peak of 550 nm is a little stronger.Our calculated results are in good agreement with the experimental results.In addition,the influence of the impurity on emission spectra is also confirmed theoretically, which adds to the understanding of the luminescent properties of TADF molecules in aggregation.
Data availability statement
The data that support the findings of this study are openly available in Science Data Bank at https://doi.org/10.57760/sciencedb.j00113.00093.
Acknowledgements
Project supported by the National Natural Science Foundation of China(Grant Nos.11974216, 11874242, 21933002 and 11904210) and Shandong Provincial Natural Science Foundation, China (Grant No.ZR2019MA056).The authors acknowledge the support of the Taishan Scholar Project of Shandong Province and the project funded by China Postdoctoral Science Foundation(Grant No.2018M642689).
猜你喜欢
杂志排行

Chinese Physics B的其它文章
- Single-qubit quantum classifier based on gradient-free optimization algorithm
- Mode dynamics of Bose-Einstein condensates in a single-well potential
- A quantum algorithm for Toeplitz matrix-vector multiplication
- Non-Gaussian approach: Withstanding loss and noise of multi-scattering underwater channel for continuous-variable quantum teleportation
- Trajectory equation of a lump before and after collision with other waves for generalized Hirota-Satsuma-Ito equation
- Detection of healthy and pathological heartbeat dynamics in ECG signals using multivariate recurrence networks with multiple scale factors