长效CRISPR/Cas9基因编辑结局的动态追踪研究
2023-10-29周雨潇吴海波
张 硕,周雨潇,吴海波,索 伦
(上海交通大学医学院附属第九人民医院,上海 200011)
CIRSPR/Cas9基因编辑技术自诞生以来,因其设计简便、操作简单等诸多优点,先后被应用于畜牧业的各个领域,如改良家畜性状、生产无过敏原的动物源性产品、动物福利优化以及生产含外源基因的转基因动物等[1]。CRISPR/Cas9的有效性分别在细胞系[2-3]、小鼠[4]、猪[5-7]、牛[8-9]以及家禽[10]上均已获得验证,为遗传育种、宿主改造、构建疾病模型和疾病治疗等的研究提供有效的技术手段[11]。
CRISPR/Cas系统发现于大肠杆菌[12-14],包括3种主要类型,目前广泛应用的CRISPR/Cas9系统是在Ⅱ型系统的基础上进行改造而来[14]。CRISPR/Cas9主要作用过程为:在sgRNA(small guide RNA,sgRNA)的指导下,Cas9蛋白对与其结合的靶向序列进行切割,导致DNA双链断裂(double strand breaking,DSB),断裂的DNA可以通过同源重组修复[15](homologous directed repair,HDR)或者非同源末端连接[16](non-homologous end joining,NHEJ)两条途径进行修复,从而实现基因改造的目的。
研究发现,CRISPR/Cas9系统不仅可以在鸡胚中稳定表达编辑[17],还可以在猪、绵羊、牛等大型动物上表达编辑[18]。截至目前,Shi等[19]使用CRISPR/Cas9技术对BMP15外显子1号区域进行编辑,获得了杂合子母猪,并展现出较高的繁殖性能,为定向育种提供了有效的实施方法;Whitworth等[20-21]使用该技术成功生产出抗PRRSV(porcine reproductive and respiratory syndrome virus)的基因编辑猪,大大提高了猪抗病能力,Ikeda等[22]运用CRISPR/Cas9技术和体细胞核移植技术成功获得IARS致病基因被纠正的胎牛,进而降低了牛感染的机率,因此,CRISPR/Cas9技术在抗病育种方面展示出巨大的优势。此外,CRISPR/Cas9技术的出现为改善畜牧业相关产品的品质提供了有效的技术手段,Wang等[23-24]成功将二花脸猪上的MSTN基因进行突变,获得双肌表型,使得猪的肌肉更加丰满,Oishi等[25]使用CRISPR/Cas系统敲除了蛋清中卵清蛋白和卵黏蛋白的基因,进而去除鸡蛋中的过敏原。
然而,目前CRISPR/Cas9的生殖系导入方式主要是采用胚胎显微注射的方式进行,对动物的胚胎生物技术要求较高[26],而某些家畜生殖细胞体外操作技术并不成熟[27],这严重阻碍了该技术的应用。业界提出通过病毒介导的方式进行在体生殖系的长效基因编辑,但长效CRISPR系统作用下,基因编辑动态结局为何,长效编辑是否会增加脱靶风险目前尚未可知。
为此,本研究借助慢病毒整合系统,先后将Cas9基因序列和靶基因序列整合到U-2 OS细胞基因组中进而实现对基因组长期稳定编辑的目的,并选择FANCF和VEGFA基因为研究对象,分析在CRISPR/Cas9基因编辑工具长期作用下靶点序列编辑效率和修复的动态结局,比较靶点序列位置对CRISPR/Cas9编辑效率和修复结局的影响,追踪CRISPR/Cas9长效作用下FANCF靶点的脱靶效应,为CRISPR/Cas9基因编辑技术在转基因动物育种的应用提供数据参考。
1 材料与方法
1.1 材料
1.1.1 试验材料 本试验使用的感受态细胞为Trans-T1菌株,购自北京全式金生物技术有限公司;U-2 OS细胞购自中国科学院上海细胞库;Cas9诱导表达质粒Tet on-Cas9-Neo-TLCV2、sgRNA质粒U6-sgRNA-TLCV2-Puromycin与慢病毒包装质粒(PsPAX和PMD2.G)均由上海交通大学中心实验室赠予。
1.1.2 试剂与试剂盒 本试验分子克隆所需的T4 DNA连接酶、限制性内切酶、DNA高保真聚合酶均购自NEB公司;质粒中量小提试剂盒购自天根生化有限公司;DMEM细胞培养基、青霉素-链霉素溶液、胎牛血清均购自Invitrogen公司;LipofectamineTM3000转染试剂购自Thermo Fisher Scientific;DNA抽提试剂盒(DNeasy Blood &Tissue Kits)购自QIAGEN公司;TruePrep Index Kit V2 for Illumina购自诺维赞生物科技有限公司;Sure PAGE预制胶购自金斯瑞生物科技有限公司;抗体试剂购自Cell Signaling Technology公司和Invitrogen公司。
1.1.3 主要仪器 PCR仪购自Applied Biosystem;高速离心机购自Eppendorf,核酸蛋白定量仪为Nanodrop 2000购自Thermo Fisher Scientific。
1.1.4 生物合成及测序服务 试验所需的各类引物合成(sgRNA寡核苷酸引物、PCR相关引物等)及后续的一代测序、二代测序均由生工生物工程有限公司完成。
1.2 方法
1.2.1 靶点表达载体构建 本试验所用2个靶区位点(FANCF和VEGFA)sgRNA克隆方法:1)使用引物(表1)以U6-sgRNA-TLCV2-Puromycin载体为模板进行PCR:①使用引物Vector primer F&R扩增TLCV2载体骨架;②使用引物scaffold-CMV primer F&R扩增载体启动子区域;③使用引物FANCFsite primer F&R或VEGFAsite primer F&R将基因对应的sgRNA片段连入载体启动子区域;2)使用NEBuilder®HiFi DNA Assembly Master Mix将载体骨架与sgRNA启动子区域进行连接,以构建完整的靶点病毒表达载体;3)使用Trans1-T1感受态完成转化过程并过夜培养;4)挑取克隆进行测序,并选择测序结果正确的克隆进行扩增,去内毒素质粒中量小提后以用于后续的细胞转染试验。

表1 引物信息
1.2.2 慢病毒包装 HEK293T细胞使用含10% FBS、100 μg·mL-1青霉素-链霉素的DMEM高糖培养基进行培养。使用LipofectamineTM3000转染试剂分别将Tet on-Cas9-Neo-TLCV2质粒、U6-FANCFsgRNA-TLCV2-Puromycin质粒、U6-VEGFAsgRNA-TLCV2-Puromycin与病毒包装质粒PsPAX2、PMD2.G共转染进HEK293T细胞,在37 ℃、5% CO2细胞培养箱内培养,转染24 h后收集细胞培养液上清,使用0.45 μM滤器过滤后使用。
1.2.3 Tet on-Cas9慢病毒感染U-2 OS细胞 U-2 OS细胞使用含10% FBS、100 μg·mL-1青霉素-链霉素的DMEM高糖培养基进行培养。将U-2 OS细胞按照3×105个·孔-1的密度接种于6孔板中,在37 ℃、5% CO2细胞培养箱内培养24 h后,按照病毒滴度为MOI=0.05感染U-2 OS细胞,慢病毒感染24 h后,将细胞培养液更换为含1 mg·mL-1G418的DMEM高糖培养基培养3~5 d,得到诱导表达Cas9蛋白的U-2 OS细胞株。
1.2.4 靶点慢病毒感染Tet on-Cas9-U-2 OS细胞 将Cas9-U-2 OS细胞按照3×105个·孔-1的密度接种于6孔板中,在37 ℃、5% CO2细胞培养箱内培养24 h后,按照病毒滴度为MOI=0.05感染Cas9-U-2 OS细胞,慢病毒感染24 h后,将细胞培养液更换为含2 mg·mL-1Puromycin的DMEM高糖培养基培养3 d,得到稳定表达靶点sgRNA以及诱导表达Cas9蛋白的U-2 OS细胞株。
1.2.5 诱导Cas9蛋白表达 将U-2 OS-Cas9细胞和稳定表达靶点sgRNA-Target以及Cas9蛋白的U-2 OS稳转株分别使用含10% FBS、5% 100 μg·mL-1青霉素-链霉素以及2 μg·mL-1Doxycycline的DMEM高糖培养基进行培养,持续诱导Cas9蛋白表达,选择不同编辑时间点收集细胞,使用DNeasy Blood &Tissue Kits(QIAGEN)提取细胞基因组DNA用于后续试验。
1.2.6 蛋白免疫印迹检测蛋白表达 取2 μg·mL-1Doxycycline诱导第3天的U-2 OS-Cas9细胞,加入100 μL含有蛋白酶和磷酸酶抑制剂的RIPA裂解液,刮刀将细胞从培养皿底刮下并转移至1.5 mL离心管中,置于冰上孵育30 min后4 ℃ 12 000 r·min-1离心10 min,吸取上清,加入5×Loading Buffer混合均匀,95 ℃金属浴加热10 min使蛋白充分变性。经电泳、转膜、封闭、一抗孵育、二抗孵育、显影等步骤得到蛋白印迹图。
1.2.7 DNA测序文库构建、二代高通量测序及数据分析 针对靶点的DNA文库构建流程如下:1)①以外源性靶点为中心设计含有index序列的特异性引物OUT-NGS-F&R、FANCF-OUT-NGS-F&R、VEGFA-OUT-NGS-F&R(表1);②以内源性靶点为中心设计含有index序列的特异性引物ONT-NGS-F&R、FANCF-ONT-NGS-F&R、VEGFA-
ONT-NGS-F&R(表1);③以FANCF靶点GUIDE-SEQ预测的7个脱靶位点为中心,分别设计含有不同index序列信息的特异性引物F1.1/1.2/1.3-NGS-F&1R、F2.1/2.2/2.3-NGS-F&2R、F3.1/3.2/3.3-NGS-F&3R、F4.1/4.2/4.3-NGS-F&4R、F5.1/5.2/5.3-NGS-F&5R、F6.1/6.2/6.3-NGS-F&6R、F7.1/7.2/7.3-NGS-F&7R(表1)。使用上述引物扩增出长约200 bp的DNA片段,用于第二轮扩增的底物。PCR组分和用量:10 μL 2×Q5 Master Mix,0.5 μL primer F(10 μmol·L-1),0.5 μL primer R(10 μmol·L-1),1 μL基因组DNA模板,RNase-Free Water补齐至20 μL。PCR参数:98 ℃预变性30 s;98 ℃变性10 s,67 ℃退火30 s,72 ℃延伸10 s,重复30个循环;72 ℃彻底延伸5 min;4 ℃储存;2)第二轮PCR建库使用TruePrep Index Kit V2 for Illumina接头引物,PCR组分和用量:10 μL 2×Q5 Master Mix,0.5 μL primer P7(10 μmol·L-1),0.5 μL primer N5(10 μmol·L-1),1 μL一轮PCR产物,RNase-Free Water补齐至20 μL。PCR参数:98 ℃预变性30 s;98 ℃变性10 s,64 ℃退火30 s,72 ℃延伸10 s,重复20个循环;72 ℃彻底延伸5 min;4 ℃储存;3)PCR结束确认条带大小正确后,将所有样品混匀后吸取200 μL进行琼脂糖凝胶电泳,经胶回收纯化测定浓度后,将纯化产物送上海生工生物工程有限公司测序。
1.2.8 统计学分析 本研究试验数据先采用Excel 2019进行整理,然后使用GraphPad Prism 7.0软件分析数据并作图,所有数据以“平均值±标准差(mean±SD)”表示。采用单因素方差分析对每个外源靶点在不同时间点的基因编辑效率差异进行分析,采用双因素方差分析各基因内、外源靶点的编辑效率差异,P<0.05表示差异有统计学意义,*代表P<0.05,**代表P<0.01,***代表P<0.001,****代表P<0.000 1。
2 结 果
2.1 靶点的构建及验证
本试验针对FANCF基因的第一个外显子区域以及VEGFA基因的第二个外显子区域,各自设计一个长20 bp的靶区,并合成含有N20以及N20-CGG的特异性序列(表1),将sgRNA(N20)和Target(N20-CGG)序列连入U6-sgRNA-TCLV2-Puromycin的慢病毒表达载体中(图1A)。测序结果显示,该克隆分别含有完整的sgRNA序列和target序列(图1 B和1C),且二者由669个碱基的linker连接,将上述测序正确的sgRNA和靶点克隆用于后续试验。
此外,为了保证细胞模型中同时表达sgRNA序列和Cas9蛋白,本研究构建了诱导型Cas9蛋白表达载体,并与PsPAX2、PMD2.G慢病毒包装载体共同转染到HEK293T细胞中用于生产慢病毒感染U-2 OS细胞(图1D),蛋白免疫印迹结果表明,在Doxycycline作用浓度为2 μg·mL-1时,可成功诱导Cas9蛋白表达(图1E)。
2.2 长效CRISPR/Cas9系统不同作用时间对编辑效率的影响
本试验将上述成功构建的含FANCF、VEGFAsgRNA和其对应靶点的TLCV2慢病毒表达载体进行慢病毒包装并感染整合有Cas9的U-2 OS细胞,在Doxycycline诱导不同时间点收集细胞样品并抽提基因组DNA用于检测基因编辑效率(图2A)。
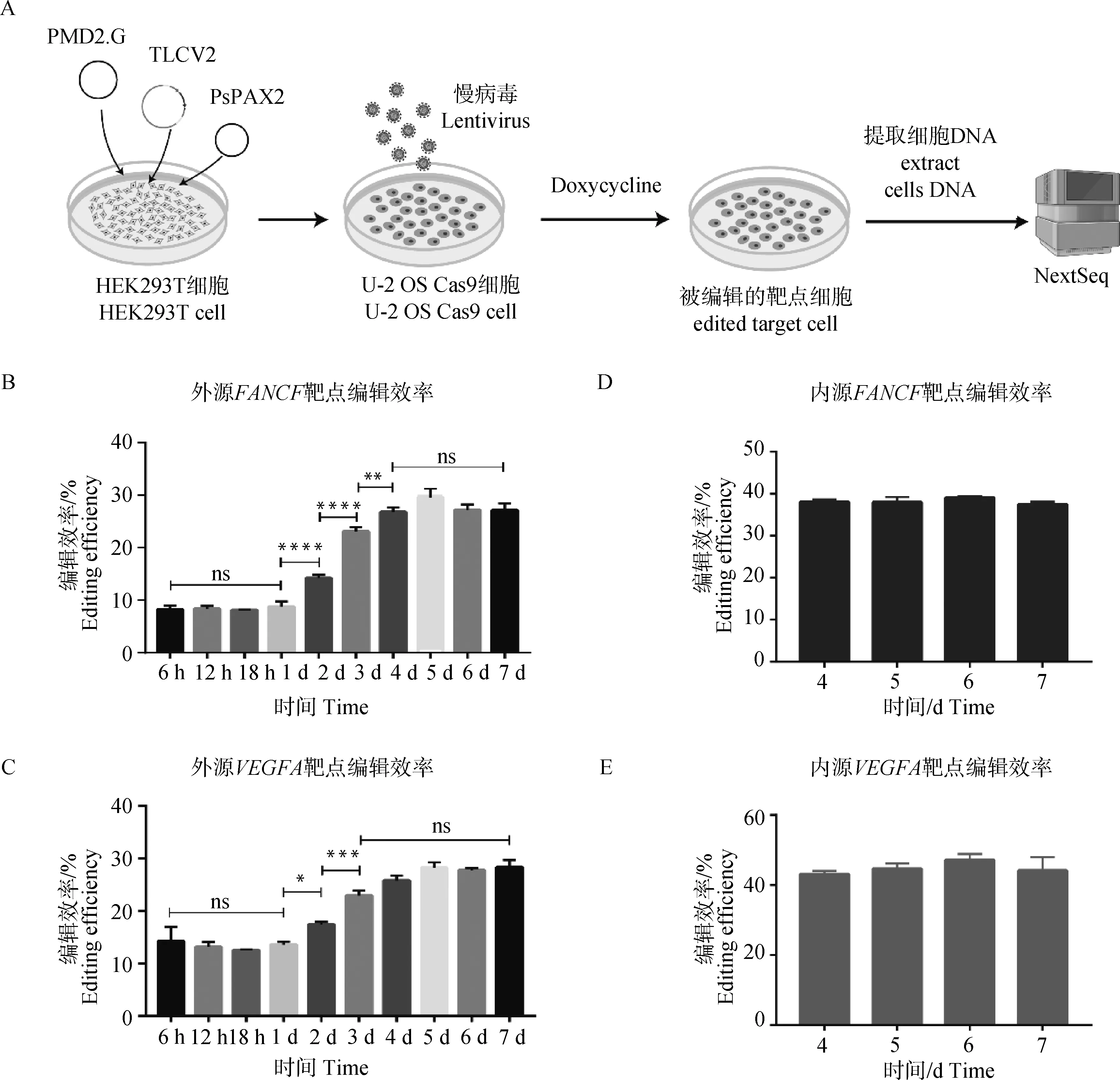
A.加入Doxycycline后使用二代测序检测细胞中外源和内源靶点区DNA的编辑情况;B.外源FANCF靶点在不同时间点的编辑效率单因素方差分析结果;C.外源VEGFA靶点在不同时间点的编辑效率单因素方差分析结果;D. 内源FANCF靶点在不同时间点的编辑效率;E.内源VEGFA靶点在不同时间点的编辑效率。*.P<0.05;**.P<0.01;***.P<0.001;****. P<0.000 1。下同A. Detection of DNA editing at exogenous and endogenous target regions in cells using next-generation sequencing following the addition of Doxycycline; B. Editing efficiency of FANCF exogenous targets at different editing times by one way ANOVA; C. Editing efficiency of VEGFA exogenous targets at different editing times by one way ANOVA; D. Editing efficiency of FANCF endogenous targets at different editing times; E. Editing efficiency of VEGFA endogenous targets at different editing times. *.P<0.05;**.P<0.01;***.P<0.001;****. P<0.000 1. The same as below图2 不同诱导时间点对CRISPR/Cas9编辑效率的影响Fig.2 Effect of different Doxycycline treatment time points on CRISPR/Cas9 gene editing efficiency
使用特异性引物分别对外源靶点和内源靶点进行PCR扩增,通过二代高通量测序技术对CRISPR/Cas9编辑后的靶点进行测序,并统计各位点编辑效率的情况,经过分析发现,FANCF外源靶点在第4天编辑效率达到峰值(图2B),VEGFA外源靶点在第3天编辑效率达到峰值(图2C);对应上述靶区的内源靶点达到峰值的时间点与外源靶点基本一致(图2D和2E)。上述结果表明,随着Cas9蛋白诱导表达时间的增加,编辑效率逐渐升高,第4天以后编辑效率逐渐趋于稳定,靶点位置对CRISPR/Cas9编辑效率的整体趋势无明显影响。
2.3 靶点位置对CRISPR/Cas9编辑效率的影响
为比较基因靶区位置对基因编辑效率的影响,本试验针对同一靶区,分别比较了内、外源靶点在第4~7天的编辑效率,结果显示编辑效率在不同时间点趋于稳定,但对于上述两个编辑靶区,其内源靶点在各时间点的编辑效率均显著高于外源靶点(图3A和3B)。
2.4 不同编辑时间点对内源基因碱基序列的影响
本试验使用特异PCR引物对内源靶点的碱基序列进行扩增并选取数据量排名前20位的结果进行分析,结果发现,内源FANCF靶点在第4~7天碱基的删除、插入、替换结局改变稳定(图4A-D),内源VEGFA靶点在编辑的各时间点碱基的删除、插入、替换结局改变也稳定(图4E-H)。上述结果说明,加入Doxycycline诱导Cas9蛋白表达在第4~7天各编辑时间点,内源靶点碱基的改变结局呈稳定状态。
2.5 不同编辑时间点对外源基因碱基序列的影响
本试验采用上述相同的方法对外源靶点的修复结局进一步分析,结果同样发现,在加入Doxycycline第4~7天,外源FANCF靶点碱基的删除、插入、替换结局改变稳定(图5A-D);在该编辑时间段,外源VEGFA靶点碱基的删除、插入、替换结局改变也稳定(图5E-H)。上述结果说明,在CRISPR/Cas9作用的第4~7天,外源靶点碱基序列的改变呈稳定状态。
2.6 靶点位置对编辑结局的影响
本试验对各基因内、外源靶点诱导7 d后编辑结局进一步比对发现,FANCF内源靶点编辑结局中插入占比为18%,删除占比为69%,其它类型占比为13%(图6A),FANCF外源靶点编辑结局中插入占比为18%,删除占比为80%,其它类型占比为2%(图6B);VEGFA内源靶点编辑结局中插入占比为13%,删除占比为85%,其它类型占比为2%(图6C),VEGFA外源靶点编辑结局中插入占比为13%,删除占比为86%,其它类型占比为1%(图6D)。上述结果说明,FANCF内源靶点和外源靶点编辑结局比重趋势一致,从大到小均依次为删除、插入、其它类型,VEGFA内源靶点和外源靶点各编辑结局占比也呈现相同的状况,表明基因靶点所处位置并不影响基因编辑后各结局占比顺序。

A.内源FANCF靶点编辑结果统计图;B.外源FANCF靶点编辑结果统计图;C.内源VEGFA靶点编辑结果统计图;D.外源VEGFA靶点编辑结果统计图A. Statistical chart of endogenous FANCF target editing results; B. Statistical chart of exogenous FANCF target editing results; C. Statistical chart of endogenous VEGFA target editing results; D. Statistical chart of exogenous VEGFA target editing results图6 靶点编辑结局统计分析Fig.6 Statistical analysis of target editing outcomes
2.7 长效CRISPR/Cas9编辑对脱靶的影响
本试验针对FANCF编辑位点,采用GUIDE-SEQ预测出6个潜在脱靶位点[28](图7),分别使用特异性引物进行PCR扩增,通过二代高通量测序技术对预测的靶点进行测序分析,各脱靶位点具体的序列信息及测序比对发现没有脱靶现象的产生。
3 讨 论
目前,CRISPR/Cas9编辑工具主要通过AAV(adneo-associated virus)、AdV(adneoviral vector)、LV(lentiviral vector)和VLP(virus-like particle)等病毒载体传递进入体内[29],进而达到长效基因编辑的目的[30-31]。研究发现,借助慢病毒载体整合到猪基因组的DNA片段在其4岁时的表达水平与其幼时相当,在羊、鸡等动物体内也观察到相似的现象,在小鼠多代传递后整合片段尚未发现明显的沉默现象[32]。目前,表达Cas9蛋白的转基因小鼠模型已用于试验研究中,通过病毒整合的方式将sgRNA整合到宿主体内即可实现对宿主任意细胞或器官的编辑[33],但长效CRISPR/Cas9提高编辑效率的同时也出现了相应的挑战,如基因编辑动物中CRISPR/Cas9的脱靶问题[34],且CRISPR/Cas9持续表达下基因编辑的动态也尚未可知。因此,为追踪长效CRISPR/Cas9作用下基因编辑的动态过程,本研究针对基因FANCF和VEGFA各自设计一个Target靶区,并将sgRNA序列和Target靶区序列构建到TLCV2质粒载体中,Cas9基因序列和靶点序列分别借助病毒载体随机整合到细胞基因组中。研究发现,在加入Doxycycline诱导Cas9蛋白表达后,对不同时间点的编辑效率和结局进行追踪分析,研究结果表明,CRISPR/Cas9持续作用的第4天之后编辑效率和结局呈现稳定状态,这为追踪长效CRISPR/Cas9基因组的编辑情况提供了有力的数据参考。
本研究通过对编辑第7天的内源靶点和外源靶点的编辑效率进行对比发现,内源靶点的编辑效率显著高于外源靶点。推测,外源靶点编辑效率显著性降低一方面可能与外源靶点整合进入基因组中的位置有关,以往研究发现通过慢病毒可以将目的序列随机整合到基因组中多个位点[35-39],在Cas9丰度相同的情况下,外源位点拷贝数远大于内源位点,导致未被编辑位点的占比提高,进而降低CRISPR/Cas9对外源位点的编辑效率[40]。在CRISPR/Cas9基因编辑的过程中,随着外源靶点整合到基因组中位置的不同,将显著影响DNA三维结构,进而影响了外源靶点的编辑效率,导致外源靶点的编辑效率低于内源靶点。另一方面可能与靶点两翼的特异性序列有关,研究表明Cas9蛋白活性受非靶点序列以外其它序列信息的影响[41],本研究中由于慢病毒载体中靶点序列的两翼序列与基因组中不同,从而导致了CRISPR/Cas9在外源靶点和内源靶点编辑效率的不同。然而,内源靶点与外源靶点编辑效率的不同并不影响对CRISPR/Cas9修复结局动态过程的追踪。通过控制Doxycycline诱导时间,进而人为控制Cas9蛋白表达并编辑基因组过程,在不同作用时间节点观察基因编辑的动态。
本研究发现,无论是FANCF位点还是VEGFA位点,通过NHEJ途径修复后,内源靶点和外源靶点删除、插入及其它类型编辑结局占比顺序一致,这一结论在Shen等[42]的研究中发现相同现象:采用相同的策略将sgRNA和target序列构建入一个载体中,对经NHEJ途径修复后内外源靶点的编辑结局中碱基删除与插入占比分析发现其顺序一致。
目前,本研究只在细胞层面研究了CRISPR/Cas9编辑及修复的动态过程,动物活体中CRISPR/Cas9编辑的具体情况尚不清晰。在未来,需要生产含有CRISPR系统的转基因动物,来进一步研究动物活体中基因编辑及修复的动态过程,进而为长效CRISPR/Cas9技术在定向育种、改良家畜性状方面的进一步应用提供更为详实的研究数据。
4 结 论
在长效CRISPR/Cas9系统作用下,靶点的编辑效率在第3或第4天达到峰值,且内外源各编辑结局类型占比趋势一致。此外,对于FANCF位点而言,CRISPR作用时间增加不会增加脱靶风险。该研究结果为动物体内追踪CRISPR/Cas9长效作用后的编辑效率和结局提供了初步的科学参考。
