市售双氯芬酸钠肠溶片质量分析与杂质风险评价
2023-10-01何积芬黄国健李沃强吕冠欣
何积芬,黄国健,李沃强,吕冠欣
1.国家药品监督管理局药物代谢研究与评价重点实验室,广东 广州 510515;2.佛山市顺德区药品检验所,广东 佛山528300
双氯芬酸钠(diclofenac sodium)又名双氯灭痛,属于第三代强效非甾体抗炎药,其作用机制是通过抑制前列腺素的合成与释放,达到减轻疼痛的目的,具有抗炎、镇痛和解热的效果[1]。市售双氯芬酸钠的口服制剂通常为肠溶制剂,以解决其酸不稳定性和对胃黏膜的刺激性,通过片芯或微丸包肠衣的方式来降低药物的不良反应[2-3]。双氯芬酸钠肠溶片以其药效强、剂量小、不良反应轻、个体差异小的优势,在骨科各类轻中度急慢性疼痛的临床治疗中应用广泛[4],是抗炎镇痛类药物的典型代表之一。为提高药品监管的有效性和针对性,本研究收集了市售29 批双氯芬酸钠肠溶片,涉及生产企业9 家。按照《中国药典》2020 年版二部[5]对样品进行检验,从溶出曲线分析不同厂家样品与原研样品的体外溶出行为的差异。同时,建立了双氯芬酸钠潜在风险杂质的定量分析方法并通过了方法学验证,用新建立的杂质定量分析方法测得的杂质个数和杂质总量大于药典有关物质测定结果,部分样品甚至出现了不合格情况,提示部分企业生产的双氯芬酸钠肠溶片存在较大质量风险,并为药典有关物质方法的改进提供了思路。
1 材料
1.1 仪器
天大天发RC-806 药物溶出仪、SOTAX AT7 smart 自动溶出仪、岛津UV-2700 紫外-可见分光光度计、赛默飞U-3000 高效液相色谱仪、梅特勒AG-204 万分之一电子天平、赛多利斯BT 125D十万分之一电子天平、上海精宏DHG-9076A 电热恒温鼓风干燥箱、上海精密WFH-203B 紫外分析仪。
1.2 药品与试剂
双氯芬酸钠对照品(中国食品药品检定研究院,批号:100334-201803,纯度:100.0%);双氯芬酸钠杂质A 对照品、杂质C 对照品、杂质E 对照品(中国食品药品检定研究院,批号分别为:100395-200301、510124-201501、510120-201501);双氯芬酸钠杂质B 对照品(QUALITY CONTROL CHEMICALS INS,批 号:06-DEC-20-02,纯 度:98.79%);29 批市售双氯芬酸钠肠溶片均为25 mg规格,涉及9 个生产厂家;甲醇为色谱纯,磷酸二氢钠、磷酸钠、氢氧化钠、冰醋酸、盐酸、磷酸、过氧化氢溶液等试剂均为分析纯,以纯化水作为实验用水。
2 方法与结果
2.1 样品概况
双氯芬酸钠肠溶片原研厂家为诺华制药,进口本地化产品由北京诺华制药有限公司生产,商品名为扶他林。自1986 年首次在我国投放市场以来,双氯芬酸钠肠溶片得到了广泛应用,生产企业众多。截至2022 年12 月,国家药品监督管理局网站(www.nmpa.gov.cn)药品数据库显示,双氯芬酸钠肠溶片有194 个药品批准文号,有25 mg 和50 mg 两个制剂规格。本次收集的29 批样品均为25 mg 规格,涵盖了9 个生产厂家[其中A 厂家(原研企业北京诺华)8 批,B 厂家8 批,C 厂家4 批,D 厂家3 批,E 厂家2 批,其余F、G、H、I 厂家各1 批],分别来自药品零售药店、连锁药店、批发企业和医疗机构,检验标准全部执行《中国药典》2020 年版二部。
2.2 依标准检验结果
双氯芬酸钠原料及肠溶片均为《中国药典》收载品种,2015 年版和2020 年版检验标准一致。依照药典标准对29 批双氯芬酸钠肠溶片进行检验,其中性状、鉴别、重量差异和含量测定等项目均符合标准规定,未发现明显的质量风险。
2.2.1 有关物质采用HPLC 自身对照法测定有关物质,标准规定相对双氯芬酸峰保留时间1.2~1.3 处的杂质Ⅲ校正后不得大于0.5%,其他单个杂质不得大于0.5%,各杂质的和(杂质Ⅲ按校正后计)不得大于1.0%。本次检验29 批样品中杂质Ⅲ校正后含量在0.001%~0.379%之间,其他单个杂质在0.018%~0.421%之间,总杂质(杂质Ⅲ按校正后计)在0.047%~0.985%之间。由于标准中未规定色谱柱型号、柱长等信息,采用不同品牌规格的C18 柱分析29 批样品,杂质Ⅲ的相对保留时间在1.22~1.36 之间。因此,采用相对保留时间来确定杂质Ⅲ,存在定位不准确的可能。同时,发现B厂家样品的总杂质含量普遍高于其他厂家,最高达到0.985%,接近标准规定的限值。方法采用等度洗脱,色谱图记录至主峰保留时间的2 倍,存在部分杂质不能完全检出的情况。因此,药典中的有关物质方法还需进一步改进,以更好地评价双氯芬酸钠肠溶片的杂质风险。
2.2.2 溶出度标准规定,双氯芬酸钠肠溶片在酸中的溶出量应不大于标示量的10%,在缓冲液中的溶出量应不小于标示量的70%。29 批样品的酸中溶出度结果均较理想,没有明显差异;缓冲液中平均溶出量B 厂家为103.4%,A 厂家为100.5%,C 厂家为97.3%。与A 厂家原研药相比,B 厂家的平均溶出量明显偏高,且同一批6 片溶出量的RSD%在0.5%~7.6%之间,不同批次间的重复性差异较大,表明该厂家产品质量存在不稳定的情况。
仿制药企业的检验结果在有关物质和溶出量两个项目上与原研企业呈现较大差异,且存在同一企业不同批次的产品质量不稳定的情况,部分甚至接近标准规定的限值。为深入分析双氯芬酸钠肠溶片的质量风险,对9 个厂家的样品从溶出曲线和杂质定量分析两方面进行了探索性研究。
2.3 溶出曲线分析
2.3.1 方法的选择根据《口服固体制剂溶出度试验技术指导原则》《普通口服固体制剂溶出曲线测定与比较指导原则》的规定,应至少选择3 种pH值的溶出介质进行溶出曲线考察[6-7]。但是,考虑到双氯芬酸钠的溶解度呈现pH 依赖性,在酸性介质中溶解降低;肠溶包衣在水中120 min 不被破损,导致主成分无法溶出;制剂不经酸性介质阶段,在pH6.8 的磷酸盐介质中的溶出速度更快等特点[8-9],最终选择经0.1 mol/L 盐酸溶液试验2 h 后,以1 000 mL 磷酸盐缓冲液(pH 6.8)作为溶出介质,转速为100 r/min,经5、10、15、20、30、45 和60 min 时,取溶液10 mL 并即时补充相同体积的溶出介质,照紫外-可见分光光度法测定吸光度,波长选择276 nm,按外标法计算每片的溶出量。
2.3.2 结果分析9 家企业的样品照上述方法试验,绘制溶出曲线,各企业溶出曲线与A 厂家生产的原研药比较见图1。由试验结果可知,双氯芬酸钠肠溶片在pH 6.8 的磷酸盐缓冲液中,存在时间为5 min~10 min 不等的溶出滞后现象。样品在第一个时间点的溶出度RSD 达到20%以上,第二个时间点的溶出度RSD 超过10%,导致溶出曲线与非模型依赖相似因子(f2)法的要求不相符。A 厂家原研药在15 min 的溶出度大于85%,可认为药物制剂的生物利用度不受溶出行为的限制,即制剂的行为与溶液相应,而仿制药溶出度达到85%的时间在5 min 至45 min 不等,尤其是B 厂家与F 厂家样品的溶出行为与A 厂家样品存在较大差异。

图1 样品溶出曲线图
2.4 杂质定量分析
《中国药典》标准对杂质Ⅲ的限值做了单独规定,双氯芬酸钠杂质B 即为《中国药典》标准中的杂质Ⅲ。有研究利用DEREK 的基因毒性警示结构来分析杂质B 的危害,认为杂质B 属于警示结构中的醛类物质,可能与DNA 上的氨基产生加成反应,从而对DNA 存在潜在的烷基化影响[10-12],因此该杂质值得我们着重关注。上述研究又采用MTT 法对各杂质的细胞毒性IC50值进行测定,结果显示杂质B 的毒性最高,进一步印证了基因毒性的预测结果。该研究对杂质进行溯源分析,发现杂质B 是双氯芬酸钠制剂在水分或光照等因素的影响下,氧化歧化降解的产物。因此,本研究针对潜在风险物质杂质B 建立了定量测定方法。
2.4.1 色谱条件色谱柱:Agilent ZORBAX Eclipse plus-C8 柱(250 mm×4.6 mm,5 μm);柱温:40 ℃;以甲醇为流动相A,磷酸盐缓冲液(称取0.8 g 磷酸二氢钠和0.5 g 磷酸,加水1 000 mL 溶解,用磷酸调节pH 值至2.5)为流动相B,梯度洗脱程序见表1;流速为1.0 mL/min;检测波长为254 nm。

表1 梯度洗脱程序
2.4.2 溶液的制备将双氯芬酸钠肠溶片研细,取约相当于双氯芬酸钠50 mg 的细粉,精密称定,置50 mL 量瓶中,加70%甲醇适量,超声使主成分溶解,用上述溶剂稀释至刻度,摇匀,离心,取上清液作为供试品溶液。
取双氯芬酸钠杂质B 对照品,精密称定,以70%甲醇为溶剂,超声溶解后,定量稀释成浓度约为1 μg/mL 的对照品溶液。
2.4.3 方法学验证(1)专属性。分别取双氯芬酸钠对照品和杂质A、B、C、E 对照品适量,精密称定,用70%甲醇溶解于同一量瓶中,并稀释制成每1 mL中各约含10 μg 的混合溶液,作为系统适用性溶液。取20 μL 注入液相色谱仪,结果见图2A,双氯芬酸钠及各杂质峰之间符合完全分离的要求。称取约相当于双氯芬酸钠50 mg 的样品细粉5 份,分别置于50 mL 量瓶中,制备破坏实验样品:①加入1 mol/L盐酸溶液2 mL,放置2 h,加1 mol/L 的氢氧化钠溶液2 mL;②加入1 mol/L 的氢氧化钠溶液2 mL,放置2 h,加1 mol/L 的盐酸溶液2 mL;③加30%过氧化氢2 mL,放置2 h;④置150 ℃加热4 h;⑤置254 nm 紫外灯下照射3 h。样品经上述破坏实验后,加70%甲醇溶解并稀释至刻度,离心,取上清液分别作为酸破坏、碱破坏、氧化破坏、热破坏以及光照破坏的样品溶液。分别取上述溶液20 μL 进样分析,结果表明产生的降解杂质均能与主成分峰较好分离,见图2B~F,本方法的专属性良好。
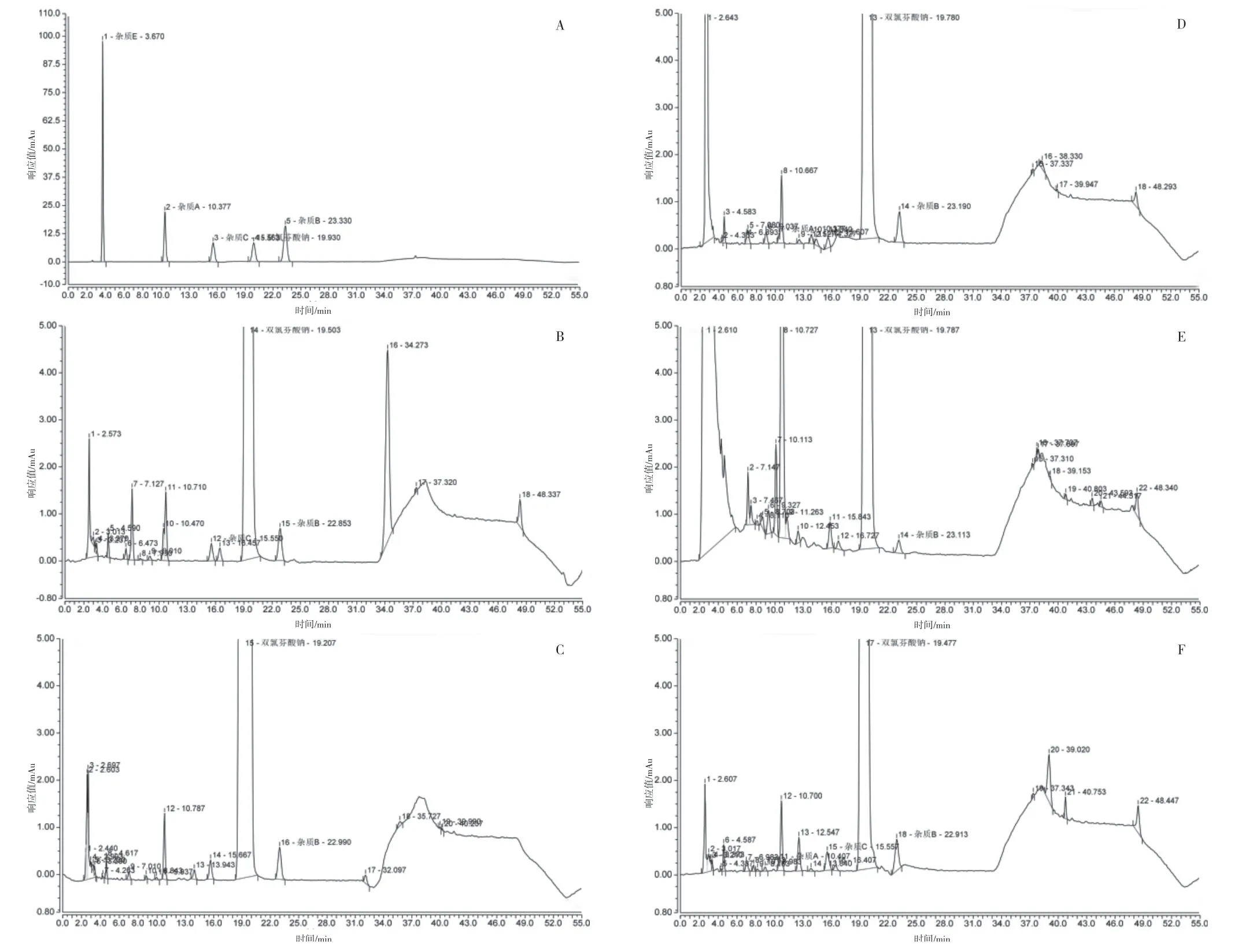
图2 专属性考察HPLC图:A.系统适用性;B.酸破坏;C.碱破坏;D.氧化破坏;E.热破坏;F.光照破坏
(2)线性关系和检出限(LOD)。精密称取双氯芬酸钠杂质B 对照品10.27 mg,用70%甲醇溶解并分别稀释制成浓度分别为0.10、0.51、1.01、2.03、5.07、20.29 μg/mL 的系列标准溶液,按“2.4.1”项下色谱条件测定,以对照品溶液的浓度作为横坐标(X),峰面积作为纵坐标(Y),绘制标准曲线,计算回归方程为Y=0.736 4X-0.017 8(r=0.999 9)。结果表明,在0.10~20.29 μg/mL 范围内,杂质B 的浓度与峰面积呈良好的线性关系。取上述标准溶液适量,用70%甲醇稀释制成0.03 μg/mL 的检出限溶液,测得色谱峰的信噪比为5.6,因此杂质B 的检出限为0.03 μg/mL。
(3)准确度。取双氯芬酸钠杂质B 对照品适量,精密称定,用70%甲醇制成标准储备液。称取样品适量,照“2.4.2”项下方法分别制备供试品溶液6 份,加入杂质B 储备液适量,使加入的对照品浓度约为1.0 μg/mL,分别进样分析,计算回收率和RSD,结果见表2。

表2 准确度考察结果
(4)重复性。取A 厂家样品,照“2.4.2”项下供试品溶液制备方法,平行制备6 份供试品溶液,照“2.4.1”项下的色谱条件进样分析,测得杂质B平均含量为71.5 μg/g,RSD 为0.8%,表明方法重复性良好。
(5)供试品溶液稳定性。取供试品溶液,于0、4、8、12、24 h 分别进样分析,杂质B 峰面积的RSD 为1.5%,表明供试品溶液中杂质B 在24 h内稳定。
3 讨论
目前该品种法定检验标准中有关物质项采用等度洗脱方法且未明确色谱型号、柱长等信息,以相对保留时间定位杂质Ⅲ,采用自身对照法计算含量,对不同厂家双氯芬酸钠肠溶片的质量区分力不强。杂质B 具有潜在的毒性风险,建议采用外标法以杂质对照品对其进行定性和定量测定。将本研究建立的杂质B 定量测定方法应用于双氯芬酸钠肠溶片有关物质分析,发现新建立的方法采用梯度洗脱且分析时间更长,对各杂质有更好的分离,各厂家样品中检出的杂质个数均多于《中国药典》法定检验方法测得的杂质个数。此外,B 厂家的部分样品用《中国药典》方法测得的总杂质含量虽然合格,但是接近标准规定的限值,最高达到0.985%。而用新建立的杂质定量测定方法测得的总杂质量大于标准规定限值1.0%,最高达到1.278%。对比两种测定方法,B 厂家8 批样品以自身对照法计算的总杂质含量,见表3。结果进一步提示了部分厂家样品的质量风险和有关物质测定方法优化的必要性。

表3 B厂家8批样品总杂质含量(自身对照法)
4 结论
本研究从溶出度和有关物质两方面考察了市售双氯芬酸钠肠溶片仿制药与原研药的质量差异,并对杂质风险进行了评估。研究结果显示多个厂家的双氯芬酸钠肠溶片与原研药的溶出行为存在较大差异。体外溶出度可预测药物的体内行为,是口服固体制剂的重要评价指标。若仿制药的体外溶出行为与原研药差异较大,则可能会对疗效的一致性产生影响,存在一定的质量风险。部分企业样品的总杂质含量偏高且接近标准限值;同一企业不同批次产品的杂质种类和含量也呈现差异。建议对现行《中国药典》双氯芬酸钠肠溶片标准中的有关物质方法进行优化,采用梯度洗脱方法,以外标法对潜在毒性杂质进行严格控制,以提高对药品的质量标准,促进企业优化制剂工艺,确保药品的安全性。
