Ⅰ型人类免疫缺陷病毒潜伏激活剂的研究进展
2023-08-01张颐娜梁瑞英田士军王娟王春颖霍珊珊于飞
张颐娜,梁瑞英,田士军,王娟,2,王春颖,霍珊珊,2,于飞,2
(1.河北农业大学生命科学学院,河北 保定 071001;2.河北省人畜共患病原微生物分析与防控重点实验室,河北 保定 071001;3.保定市妇幼保健院,河北 保定 071066)
艾滋病(acquired immune deficiency syndrome,AIDS)是一种由人类免疫缺陷病毒(human immunodeficiency virus,HIV)感染引起的危害性极大的传染病。HIV 可攻击人体的免疫系统,主要感染人体免疫系统中最重要的CD4+T淋巴细胞,使人体丧失免疫功能。HIV 可分为Ⅰ型HIV(HIV-1)和HIV-2 2 个主要类型。HIV-1 是最常见和最广泛传播的类型,是导致全球AIDS 疫情的主要病毒株;HIV-2 是一种较罕见的类型,主要在西部非洲地区流行。与HIV-1相比,HIV-2感染的进展速度较慢,病程较长,且对抗逆转录病毒药物的抵抗性较高。AIDS 已成为严重的公共卫生和社会问题,但世界范围内仍缺乏根治HIV-1感染的有效药物。现阶段的治疗目标是最大限度和持久抑制患者体内的病毒复制,重建并有效维持患者的免疫功能,降低HIV-1 感染和非AIDS 相关疾病的发病率和死亡率。感染早期应用高效抗逆转录病毒治疗(highly active antiretroviral therapy,HAART)可有效抑制病毒复制,控制病情进展。然而,单纯采取HAART 不能彻底清除患者体内的HIV-1,必须终生用药,一旦停药,潜伏病毒库会很快被激活并再度出现新一轮感染。长期用药不仅经济负担重,也面临病毒耐药、变异和药物毒性积累等问题。因此,急需开发新的创新性方案实现AIDS的功能性治愈。2012年,Deeks[1]首次提出了“shock and kill”策略,该策略利用HIV-1潜伏激活剂(latency-reversing agents,LRA)激活病毒潜伏库,然后将HIV-1 LRA 与HAART 联合使用,阻止活化病毒进一步感染,并结合增强自身免疫系统或使用抗病毒药物逐渐清除病毒潜伏库,最终实现对AIDS 的功能性治愈。目前尚无LRA 或LRA 组合药物能够彻底激活病毒潜伏库,本文综述HIV-1的潜伏机制和各类LRA 代表性药物及作用机制,以期为LRA的研发提供思路。
1 HlV-1的潜伏机制
HIV-1 属于逆转录RNA 病毒,其基因组是由2 个拷贝的单股正链RNA 组成,它们通过1 个帽状结构在5′端形成二聚体。HIV-1基因组的两端各有1个长末端重复序列(long terminal repeats,LTR),包含启动子、增强子和负调控区。LTR 之间的序列编码多种蛋白质,包括结构蛋白(Gag,Pol和Env)、调节蛋白(Tat,Rev 和Nef)和辅助蛋白(Vif,Vpr 和Vpu)[2]。
早期转录产物主要是多剪接的短转录本,合成Tat和Rev等多种调控蛋白,是HIV-1早期感染的必需的蛋白。长mRNA 包括未剪接mRNA 和单剪接mRNA,未剪接mRNA 翻译成Gag 和Pol 等多种结构蛋白,并可用作子代病毒的基因组,不完全剪接或单剪接mRNA 翻译成病毒辅助蛋白Vif,Vpr 和Vpu 等,均是HIV-1 感染晚期所必需的蛋白质。在HAART 研究中,大多数患者的CD4+T 细胞中只能检测到较少未剪接mRNA,其中大多都是不完整转录本,表明潜伏感染细胞中的HIV-1 转录效率较低[3]。短转录本出现频率较高,而完全转录本和多聚腺苷酸化转录本出现频率较低[4]。因此,LRA 主要针对提高HIV-1的转录水平。
目前HIV-1潜伏库的形成与维持的相关分子机制主要有5 种:①HIV-1 基因表达的表观遗传学调控。HIV-1 基因通过可逆的染色质修饰,如组蛋白甲基化、乙酰化和去乙酰化等,诱导染色质重塑,影响染色体的凝聚状态,并决定转录因子对基因的调控[5]。HIV-1 的转录起始位点和转录调控序列位于LTR 区域,转录调控序列是特异性转录因子的识别区域。在静息CD4+T 细胞中,调控因子与LTR 结合,通过募集组蛋白去乙酰化酶(histone deacetylase,HDAC)将调控序列中的组蛋白去乙酰化,使其被重塑为一种致密结构,抑制RNA 聚合酶Ⅱ(RNA polymerase Ⅱ,RNAPⅡ)与启动子的结合,从而阻止HIV-1基因的转录起始。同时调控因子还可通过募集组蛋白甲基转移酶(histone methyltransferase,HMT)使调控序列中的组蛋白甲基化,从而导致正常染色体异染色质化,或通过募集DNA 甲基转移酶(DNA methyltransferase,DNMT)来甲基化LTR 区域的CpG 岛,抑制基因转录,促使HIV-1 潜伏。最终大量凝集的病毒异染色质在CD4+T 细胞中累积,形成HIV-1 潜伏库。②T 细胞内活化转录因子对基因的调控。T 细胞核中的活化转录因子与潜伏密切相关。HIV-1 基因转录的启动子位于5′LTR 上,包含多个转录因子结合位点[6]。这些转录因子包括NF-κB、活化的T 细胞核因子(nuclear factor of activated T cells,NFAT)、激活蛋白1(activator protein 1,AP-1)和Tat 等。静息状态下,NF-κB 游离在细胞质中与NF-κB 抑制蛋白(inhibitor of NF-κB,IκB)结合,无法进入细胞核,活化后的NF-κB 与HIV-1 LTR 上的κB 元件结合,在HIV-1 基因的转录中起关键作用。此外,正性转录延伸因子b(positive transcription elongation factor b,P-TEFb)可被Tat招募到HIV-1 基因的启动子,通过解旋调控序列并推动磷酸化的RNAP Ⅱ向前移动来促进病毒基因的转录。静息CD4+T 细胞中仅存在少量活化转录因子,是形成HIV-1 潜伏的主要因素。③免疫信号通路的调节。通过激活CD4+T 细胞中的免疫信号通路如Janus 激酶/信号转导和转录激活子(janus kinase/signal transducers and activators of transcription,JAK/STAT)通路,可同时起到激活潜伏病毒和免疫治疗的作用。部分LRA 既能激活潜伏的HIV-1,又能增强细胞的免疫功能,从而提高感染细胞的病毒清除率。Toll 样受体(Toll like receptor,TLR)激活剂既可逆转HIV-1的潜伏状态,又可导致树突状细胞(dendritic cells,DC)成熟、自然杀伤(natural killer,NK)细胞活化、抗原呈递效率提高和适应性免疫应答增强[7]。④前病毒基因整合位点的影响。研究发现,前病毒基因大多整合在活化的mRNA 内含子中[8]。若前病毒基因整合到宿主染色体的异染色质区或基因间隔区,其染色质环境不利于HIV-1基因转录,则易发生潜伏。⑤微RNA(microRNA,miRNA)对HIV-1基因表达的影响。miRNA 是一种小的单链非编码RNA,通过与Nef基因或HIV-1 mRNA 的3′端相互作用来阻碍HIV-1基因转录,抑制其基因表达,促进其潜伏[9]。
2 HlV-1潜伏激活剂
HIV-1 LRA主要包括以下3类10种:①表观遗传修饰剂,包括HDAC 抑制剂、DNMT 抑制剂和HMT 抑制剂;②转录因子调节剂,包括蛋白激酶C(protein kinase C,PKC)激活剂、P-TEFb 激活剂、溴结构域和超末端结构域(bromodomain and extra terminal domain,BET)抑制剂、第二线粒体源胱天蛋白酶激活物(second mitochondria-derived activator of caspases,SMAC)类似物;③免疫激活剂,包括TLR 激活剂、细胞因子(cytokine,CK)和免疫检查点(immune checkpoint,IC)抑制剂等。HIV-1 潜伏激活剂的分类、代表性化合物及其作用机制见表1,代表性化合物结构见图1。
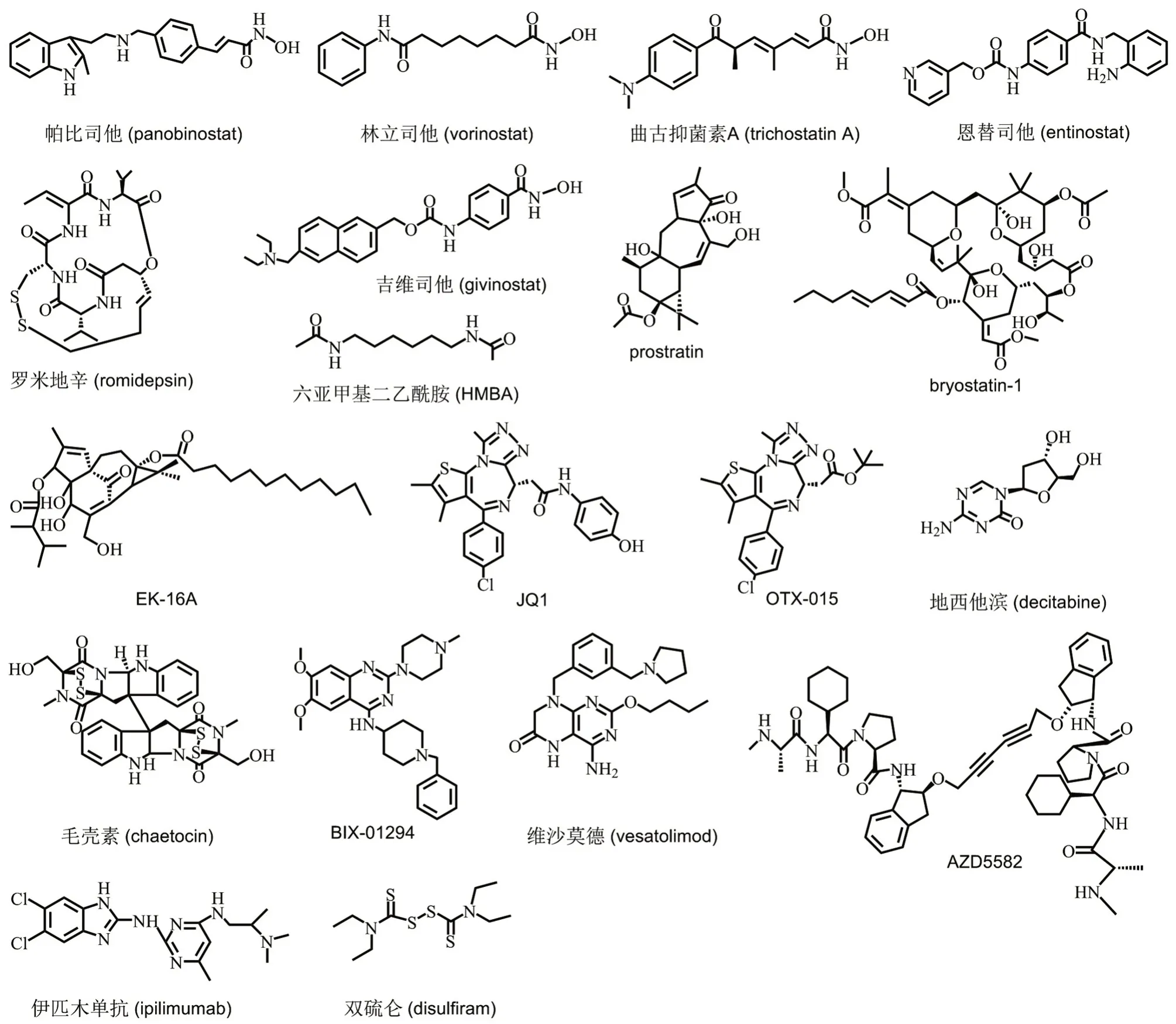
图1 HlV-1潜伏激活剂的主要代表性化合物结构.

表1 HlV-1潜伏激活剂的分类、代表性化合物及其作用机制
2.1 HDAC抑制剂
HDAC 是一类与表观遗传修饰相关的蛋白酶,对染色体的结构修饰和基因表达调控发挥重要作用。HDAC 可使组蛋白去乙酰化,使带正电荷的组蛋白与带负电荷的DNA紧密结合,染色质呈致密卷曲状态,基因的转录受到抑制。一般情况下,组蛋白的乙酰化有利于DNA与组蛋白八聚体的解离,核小体结构松弛,从而使各种转录因子和协同转录因子能与DNA 结合位点特异性结合,激活基因的转录。在细胞核内,组蛋白乙酰化与去乙酰化过程处于动态平衡,并由组蛋白乙酰化转移酶(histone acetyltransferase,HAT)和HDAC 共同调控。HAT将乙酰辅酶A的乙酰基转移到组蛋白N端特定的赖氨酸残基上。HDAC 有HDACⅠ~Ⅳ4 种类型,其中Ⅰ类HDAC 在HIV-1 潜伏中起重要作用[75]。选择性HDAC 抑制剂可能只激活部分病毒潜伏库,而不会彻底激活CD4+T 细胞中的潜伏病毒。HDAC抑制剂通过形成自噬小体抑制HIV-1在人巨噬细胞间的传播,并诱导巨噬细胞中HIV-1 DNA 的降解[76]。此外,HDAC 抑制剂还下调HIV-1 共受体的表达。
HDAC 抑制剂通过抑制HDAC 活性,减少组蛋白去乙酰化,从而促进HIV-1的转录,激活病毒潜伏库。目前,HDAC抑制剂是临床上最有效的一类LRA。
2.1.1 帕比司他(panobinostat)
帕比司他是一种有效的口服广谱性HDAC 抑制剂,8~31 nmol·L-1即可诱导HIV-1 激活,且在远低于临床治疗剂量下,亦可激活CD4+T细胞中潜伏的HIV-1[10]。帕比司他激活效率高,但细胞毒性也较大[11]。研究显示,将病毒潜伏库重复暴露于帕比司他中,能有效扰乱HIV-1 在体内的潜伏[12]。在帕比司他治疗期间,细胞内未剪接的HIV-1 mRNA 水平较基线显著升高,最高升幅达3.5 倍,但并未引起潜伏感染细胞数量减少,可能需要与其他药物相结合才能有效减少HIV-1病毒潜伏库[12]。也未观察到HIV-1感染者体内基因表达模式的长期变化[13]。在癌症治疗中,纳米药物递送体系可通过特异性靶向细胞将转运分子释放到特定细胞器等方法可克服传统药物递送的局限性。利用纳米技术,提高HDAC 抑制剂对潜伏HIV-1 的激活效率。研究表明,包裹帕比司他的纳米粒子的激活效率优于单用帕比司他,且无毒性作用,并具有良好生物相容性[14]。由于较高剂量帕比司他具有毒性,使得优化给药方式非常必要,如通过纳米技术等改进药物对其靶标部位的输送,可提高LRA的效力。
2.1.2 林立司他(vorinostat)
林立司他是一种含羟肟酸结构的Ⅰ和Ⅱ类HDAC 抑制剂,其安全性高,耐受性好,对HDAC1和HDAC3 的IC50值分别为10 和20 nmol·L-1。用从低水平病毒血症患者体内分离出的静息CD4+T细胞研究林立司他在病毒潜伏库中的作用显示,接受林立司他处理的CD4+T 细胞中HIV-1 RNA 水平增加4.8倍;与基线RNA水平相比,林立司他400 mg单次给药后,HIV-1 RNA 表达量显著增加[15]。林立司他并未促进HIV-1 特异性T 细胞增多或免疫激活,但却引起血液中调节性T 细胞数量增加。在58%的患者CD4+T 细胞中,林立司他诱导HIV-1 RNA 水平持续增加,但并不引起血浆中HIV-1 RNA表达量的改变,也未引起潜伏感染细胞频率的改变,因此需要更多有效的干预措施来诱导HIV-1 的激活,并最终消除被感染细胞[16],这些表明林立司他激活效率低,单用不能完全激活HIV-1潜伏库。
2.1.3 曲古抑菌素A(trichostatin A)
曲古抑菌素A是一种链霉菌代谢产物,是Ⅰ类和Ⅱ类HDAC抑制剂,对HDAC的IC50值为1.8 nmol·L-1。其可延缓IκB 的胞内再生,促进活化的NF-κB 与LTR 上的κB 元件结合,促进病毒转录[17]。HIV-1 转录依赖染色质上少量有转录活性的模板,每个模板可支持近100 轮转录。研究发现,曲古抑菌素A 可增加每轮活性模板的数量[18]。曲古抑菌素A处理可抑制细胞周期,是一种潜在的抗癌药物。Apicidin是一种广泛使用的抗寄生虫药,研究表明,Apicidin 可协同曲古抑菌素A 激活HIV-1 基因的转录[19]。此外,曲古抑菌素A 与肿瘤坏死因子(tumor necrosis factor,TNF)可协同激活HIV-1 LTR转录,二者间的协同作用严格依赖于靶基因上特定的DNA 序列(κB位点)的存在[20]。
2.1.4 恩替司他(entinostat)
恩替司他是一种苯甲酰胺结构的Ⅰ类HADC抑制剂,其对HDAC1 和HDAC3 的IC50分别为0.51和1.7 μmol·L-1。恩替司他可诱导潜伏感染的原代T 细胞产生病毒,其效力与林立司他相似[11]。它不增加T 细胞表面受体趋化因子受体4(C-X-C chemokine receptor type 4,CXCR4)和趋化因子受体5(C-C chemokine receptor type 5,CCR5)的表达量,所以不提高HIV-1激活后入侵细胞的风险,且毒性小于帕比司他[11]。恩替司他通过选择性阻碍Ⅰ类HDAC 与HIV-1 LTR 结合和促进组蛋白H3 乙酰化诱导病毒粒子产生[11]。将其与苔藓抑素(bryostatin-1)联用可最大限度诱导病毒蛋白表达、促进病毒粒子产生和感染细胞死亡[21]。
2.1.5 罗米地辛(romidepsin)
罗米地辛在nmol 浓度即可抑制Ⅰ类HDAC 的活性,其对HDAC1 和HDAC2 的IC50值分别为36和47 nmol·L-1,被美国FDA 批准用于治疗皮肤和外周T 细胞淋巴瘤。罗米地辛激活HIV-1 潜伏库的能力与帕比司他相近,但对CD8+T细胞具有较高的细胞毒性,平均细胞死亡率为(22±6)%[22]。研究表明,罗米地辛可安全地激活HIV-1 复制,而不改变HIV-1 特异性T 细胞的比例,也不抑制其产生CK,即使导致T 细胞受体受损,也不会减弱T 细胞介导的免疫反应[23]。罗米地辛可显著抑制HIV-1侵染外周血单核细胞(peripheral blood mononuclear cell,PBMC)和CD4+T 细胞。因此,在HAART 治疗后病毒载量下降不理想的情况下,基于罗米地辛的治疗策略可使病毒几乎不能重新形成潜伏库。
2.1.6 吉维司他(givinostat)
吉维司他主要选择性抑制Ⅰ类HDAC,其对HDAC1 和HDAC3 的IC50值分别为198 和157 nmol·L-1。吉维司他有3 大优点:①在安全使用浓度下,体外诱导潜伏感染细胞中HIV-1 的表达量至少增加10 倍,而丙戊酸作为Ⅰ类HDAC 抑制剂,诱导细胞中的HIV-1增加不到2倍;②口服给药在人体中有很好安全性(包括儿童)[10];③不增加CD4+T 细胞表面CCR5 表达,并使CD4+T 细胞表面CXCR4 和单核细胞表面CCR5 的表达量降低50%,诱导HIV-1 重新激活的比例是CD3/CD28 单克隆抗体的57%~74%。在U1 和ACH2 细胞中,吉维司他的激活能力是林立司他的50~100 倍[20]。研究表明,吉维司他与PKC联用可诱导HIV-1 RNA的表达[24]。吉维司他不仅诱导潜伏感染细胞株产生HIV-1,同时大大降低HIV-1 共受体的表达[25]。此外,吉维司他的安全使用浓度(达400 nmol·L-1)高于帕比司他和罗米地辛,这可能是其逆转HIV-1潜伏效率较强的原因之一[20]。
2.2 PKC激活剂
PKC 是G 蛋白偶联受体系统中的效应物,在非活性状态下是水溶性的,游离在胞质中。其激活依赖二酰甘油(diacylglycerol,DAG)的存在和胞质中Ca2+浓度的升高,即DAG 在质膜中出现时,PKC 从胞质中移位到质膜上,然后在Ca2+作用下被激活。
PKC 可调控基因表达,PKC 激活剂通过对PKC 信号通路的激活,激活HIV-1 潜伏库。在静息状态下,细胞中的IκB与NF-κB结合形成复合体,可使NF-κB 以无活性形式滞留在胞质中。IκB 激酶(IκB kinase,IKK)被激活后,可促进IκB 磷酸化、泛素化和随后的降解,使得IκB 释放NF-κB,然后NF-κB迅速易位入核并结合于靶基因上的κB位点,从而促进HIV-1基因转录。
2.2.1 prostratin
prostratin 发现于萨摩亚的Mamala 树的树皮中,是一种非肿瘤促进性佛波酯。prostratin 通过PKC 介导的通路活化有转录活性的NF-κB,使其进入细胞核内激活HIV-1 DNA的表达(表1)。研究表明,PKCθ、PKCε 和蛋白激酶D3(protein kinase D3,PKD3)是prostratin 诱导HIV-1 转录激活所必需。在信号传递中,PKCθ和PKCε通过磷酸化PKD3使其激活,随后活化的PKD3 通过调控NF-κB 信号途径激活HIV-1 的基因表达。综上,prostratin 诱导激活HIV-1 基因转录的信号通路为prostratin-
PKCθ/PKCε-PKD3-NF-κB-HIV-1-LTR[26]。prostratin能够激活转录起始,但对转录延伸没有明显影响[27]。prostratin 可与六亚甲基二乙酰胺(hexamethylenebisacetamide,HMBA)通过不同机制协同激活HIV-1 基因转录。prostratin 通过活化P-TEFb 从而促进HMBA 诱导的HIV-1 基因转录延伸,HMBA 通过促进prostratin 诱导的IκB 降解,使得NF-κB 易位入核及HIV-1 基因转录起始。二者共处理的激活作用可能是通过下调NF-κB 信号通路中负反馈调节因子脱泛素酶A20 的表达,致使NF-κB 信号通路长时间持续活化[28]。研究表明,prostratin与HDAC抑制剂联合使用对潜伏的HIV-1有很强的协同激活作用[26]。此外,prostratin还通过降低CD4 和CXCR4 受体表达量从而抑制HIV-1 进入靶细胞[29-30]。
2.2.2 勃利抑素1(bryostatin-1)
勃利抑素1 是从海洋生物草苔虫(Bugula neritina)中提取的一种PKC 激活剂,能持续活化PKC并最终导致PKC 耗竭[31]。勃利抑素1 通过Nef 与Tat 介导的LTR 反式激活,协同调节HIV-1 的重新激活,AMP 依赖的蛋白激酶激活PKC,进而促进HIV-1 的重新激活[32]。勃利抑素1 在nmol 浓度下即可激活潜伏的HIV-1[31],对T 细胞的安全浓度高达100 nmol·L-1[33];重新激活潜伏病毒的能力是prostratin 和林立司他的25~1000 倍。勃利抑素1与HDAC 抑制剂(如帕比司他和罗米地辛)联用可协同激活HIV-1 的基因表达[34]。勃利抑素1 还可下调HIV-1 受体CD4 和共受体CXCR4 的表达,阻止易感细胞中HIV-1 的初始感染[35]。此外,研究发现勃利抑素1与其他在医疗实践中常用的HDAC抑制剂(如VPA)有很强的协同作用[35]。
2.2.3 EK-16A
EK-16A 是一种巨大戟醇衍生物,提取自大戟属植物甘遂(Euphorbia kansuiT.N. Liou ex S. B.Ho),与prostratin 结构相似,但比prostratin 的重新激活效率更高。EK-16A 对HIV-1 的重新激活主要依赖于PKCγ。EK-16A 激活PKCγ 后,IκB 蛋白被磷酸化和泛素化降解,使NF-κB 易位入核,诱导HIV-1 LTR 转录起始;同时通过促进细胞周期蛋白依赖性激酶9(cyclin-dependent kinase 9,CDK9)磷酸化和上调细胞周期蛋白T水平活化P-TEFb,从而促进转录延伸。研究表明,在潜伏感染的Jurkat细胞中,EK-16A 可在nmol 水平显著激活HIV-1 的基因转录,活性远高于prostratin。EK-16A 在有效激活浓度(1~100 nmol·L-1)未检测到细胞毒性,且未显著增加CK 表达,虽然其使T 细胞在活化早期表达CD69,但对另一个激活标志物CD25 的促表达程度很小[36]。
2.3 P-TEFb激活剂
HIV-1 转录可分为起始、延伸和终止3 个阶段,其中延伸阶段耗时最长。P-TEFb 由CDK9 及其调节亚基周期蛋白T组成,其活性受到严格调控,维持动态平衡,在体内以活性型和非活性型2 种形式存在,当细胞处于紫外线、HMBA 或心肌肥大等刺激下,7SK 核糖核蛋白(7SK small nuclear ribonucleoprotein,7SK snRNP)复合体解离并释放有活性的P-TEFb(cyclin T/CDK9)。P-TEFb 一方面通过磷酸化苯并咪唑敏感性诱导因子和负性转录延伸因子解除其对RNAPⅡ的抑制,另一方面通过直接催化RNAPⅡC 端结构域(carboxyl-terminal domain,CTD)发生磷酸化,促使转录进入延伸阶段。溴结构域蛋白4(bromodomain-containing protein 4,BRD4)在P-TEFb由非活性型向活性型转变中发挥重要作用,还可增强P-TEFb 对RNAPⅡ的磷酸化作用。但由于BRD4 和Tat 竞争性结合P-TEFb,故常使用BET抑制剂来增强Tat和P-TEFb的结合进而激活病毒潜伏库。
2.3.1 直接激活剂HMBA
HMBA 是一种P-TEFb 激活剂,也是有效的细胞生长抑制剂和细胞分化诱导剂。HMBA 可诱导7SK snRNP 复合物的解离,从而释放有活性的P-TEFb,诱导染色质释放BRD4,通过BRD4将活性P-TEFb 重新招募到启动子上,从而刺激转录延伸。P-TEFb 的活化也受多种信号通路的调控,如通过蛋白磷酸酶2B 诱导7SK snRNP 复合体构象改变,促进CDK9上T186去磷酸化,使P-TEFb从7SK snRNP 复合物中解离出来。也可通过激活磷脂酰肌醇3 激酶/蛋白激酶B(phosphati dylinositol 3-kinase/protein kinase B,PI3K/Akt)信号通路,促使蛋白磷酸化,继而释放有活性的P-TEFb[37]。HMBA 也可能是通过非经典的NF-κB 途径激活HIV-1转录[38],诱导潜伏感染细胞系中病毒的产生。在HMBA 处理的潜伏感染细胞系中,PI3K/Akt通路的激活是产生病毒所必需[39]。HMBA 还可和prostratin 协同激活HIV-1 转录。通过阻断负反馈通路,HMBA 可作为NF-κB 途径的信号增强剂发挥作用[40]。prostratin 通过促进P-TEFb 活化来增强HMBA 诱导的HIV-1基因转录,从而促进转录延伸。HMBA 除了激活HIV-1 转录,也可下调HIV-1 表面受体表达,阻碍HIV-1感染其他细胞。
2.3.2 间接激活剂(BET抑制剂)
人BET 家族由BRD2,BRD3,BRD4 和BRDT组成,主要通过识别并结合组蛋白尾部的乙酰化赖氨酸残基,发挥调节基因转录和细胞生长的作用。人BET 家族的4 个成员有相似的结构,即2 个串联溴结构域(BD1 和BD2)及1 个超末端(ET)结构域,但生物学功能存在差异。
其中BRD4与HIV-1转录激活密切相关,BRD4末端有1 个CTD。CTD 通过募集P-TEFb 到BRD4靶基因——HIV-1 基因转录区上,促进RNAPⅡ磷酸化,进而促使转录延伸。Tat 是病毒编码的主要调控蛋白,能够使HIV-1转录水平提高千倍,它可与转录出的反式激活应答序列结合,在SF1 等细胞因子的帮助下大大加强RNAPⅡ的延伸能力,合成大量全长mRNA[77]。BRD4与Tat竞争性结合P-TEFb,从而抑制HIV-1 转录。BET 抑制剂抑制BRD4 与P-TEFb 结合,可增强Tat 与P-TEFb 的结合,提高HIV-1转录效率。
2.3.2.1 JQ1
JQ1 是一种特异性BET 抑制剂,对BRD4 的IC50通常在10~100 nmol·L-1。JQ1 通过与热休克蛋白90(heat shock protein 90,HSP90)-细胞分裂周期蛋白37(cell division cyclin,CDC37)-CDK9复合物稳定结合调控潜伏HIV-1 的转录激活。当HSP90 活性降低和CDC37 的表达量下降时,7SK snRNP 复合物重新组装,使更多CDK9 处于无活性状态,从而降低JQ1 激活潜伏HIV-1 基因表达的效率。JQ1 还可促进热休克转录因子1(heat shock transcription factor 1,HSF1)与HIV-1 LTR 区域结合,并使HSF1 募集更多HSP90-CDK9 复合物来参与其激活潜伏HIV-1基因转录的过程[41]。JQ1可能是通过从HIV-1 LTR 中移除BRD4 而使Tat 随后能与之结合[42]。JQ1还被证明在Jurkat T细胞中以非Tat依赖的方式促进HIV-1转录。研究发现,前花青素C1(procyanidin C1,丝裂原活化蛋白激酶激活剂)与PKC 激活剂和BET 抑制剂(JQ1)联合治疗时,较低浓度的3 种化合物均能有效激活潜伏的HIV-1[43]。PKC激活剂PEP005和BET抑制剂JQ1可协同激活HIV-1 潜伏储存库[44]。从虎杖(Polygonum cuspidatumSieb.et Zucc.)中提取的天然产物REJC1G3是一种P-TEFb激活剂,可通过诱导P-TEFb从7SK snRNP中释放与JQ1起协同作用[45]。
2.3.2.2 OTX-015
OTX-015是一种有效的BRD2/3/4抑制剂,IC50值为92~112 nmol·L-1。OTX-015(0.1,1,5 μmol·L-1)可诱导接受HAART 患者的静息CD4+T 细胞产生HIV-1全长转录本和病毒粒子。OTX-015通过增加CDK9 环磷酸化,促进HIV-1 LTR 对P-TEFb 的募集,进一步诱导RNAPⅡCTD 磷酸化和病毒转录,从而激活潜伏HIV-1[46]。OTX-015 对细胞活力无明显影响,既不诱导T 细胞活化,也不诱导HIV-1 受体/共受体的表达。因此,在重新激活病毒期间,OTX-015 不增加CD4+T 细胞对HIV-1 的易感性。此外,OTX-015 与prostratin 联用可有效提高激活潜伏HIV-1 的效率。但OTX-015 激活效率不高,需要优化给药浓度和时间,或与其他激活剂联合,以进一步提高其诱导HIV-1基因表达的效率。
2.4 DNMT抑制剂地西他滨(decitabine)
DNA甲基化是在DNMT作用下,将甲基选择性添加到特定碱基上。DNA 甲基化修饰是真核细胞调控基因表达的特点之一,其通过3 种途径抑制基因表达:①甲基化的CpG 岛阻碍转录因子的结合,直接抑制基因表达;②通过招募DNA 甲基结合蛋白及一些阻遏复合物,阻止转录因子和特定DNA序列结合,间接抑制基因表达;③凝集状态的染色质阻碍转录因子与其调控序列的结合。DNMT 抑制剂通过抑制病毒DNA 甲基化过程,进而促进HIV-1基因转录。一些DNMT抑制剂(如核苷类似物)磷酸化后能与DNMT 形成共价化合物,阻碍DNMT 与DNA结合,抑制其转甲基功能,诱导DNA去甲基化。
地西他滨是2-脱氧胞苷类似物,又称5Aza-CdR,高浓度时具有细胞毒作用,低浓度时具有去甲基化作用。5Aza-CdR 本身是很弱的HIV-1 LRA,但与prostratin 联用可显著提高多数J-Lat 细胞中病毒基因表达的效率[47]。研究表明,LRA 浓度及联合治疗时给药顺序对于协同激活HIV-1基因表达至关重要。5Aza-CdR+HDAC 抑制剂(恩替诺特除外)顺序给药在2 个潜伏感染的T 细胞系中协同诱导了HIV-1的基因表达[48]。最近一项在小鼠获得性免疫缺陷综合征模型中的研究结果表明,5Aza-CdR 单独或与吉西他滨(嘧啶类抗肿瘤药物)联用,通过在病毒逆转录过程中引入致命突变来抑制HIV-1的复制,导致病毒潜伏库缩小[49]。此外,在潜伏感染的Jurkat细胞和原代T细胞中的HIV-1 CpG岛上,发现甲基CPG 结合域蛋白2(methyl CpG binding domain protein 2,MBD2)结合到甲基化的胞嘧啶上,进而募集HDAC 促使组蛋白去乙酰化,MBD2和去乙酰化的组蛋白通过募集更多的DNMT来加强沉默信号,而用5Aza-CdR 抑制胞嘧啶甲基化可消除对MBD2和HDAC的募集[50]。
2.5 HMT抑制剂
组蛋白甲基化修饰一般发生在组蛋白赖氨酸和精氨酸残基上,分别由组蛋白赖氨酸甲基转移酶和组蛋白精氨酸甲基转移酶催化。在HMT 的作用下,以S-腺苷甲硫氨酸(S-adenosylme-thionine,SAM)为甲基供体,可将SAM 上的甲基转移到赖氨酸侧链的N 原子上。组蛋白赖氨酸甲基化常发生在H3K4,H3K9,H3K27和H3K36等位点上,这些位点也可发生多甲基化。组蛋白赖氨酸甲基化对基因转录起激活或抑制作用。H3K9的甲基化与基因沉默相关,而H3K4的甲基化则与基因的激活相关。组蛋白甲基转移酶SUV39可特异性甲基化H3K9。HMT抑制剂通过抑制SUV39,包括SUV39H1,SUV39H2,G9a 和ESET,抑制组蛋白H3K9甲基化,从而促进潜伏HIV-1基因的转录。
2.5.1 毛壳素(chaetocin)
毛壳素是一类来自毛壳菌属(Chaetomium kunze)的天然产物,是组蛋白甲基转移酶SUV39H1的抑制剂(表1)。研究表明,由于HIV-1 基因的表观遗传沉默限制了基因表达,因此即使在T 细胞激活的情况下,病毒仍可能处于潜伏状态[51]。毛壳素抑制H3K9的三甲基化(H3K9me3)并促进组蛋白乙酰化水平增加,引起LTR 启动子区的染色质重塑,从而产生了一个允许多种转录激活因子结合的环境,进而激活转录。90 nmol·L-1是毛壳素激活HIV-1 基因表达并保持细胞活力的最佳浓度,可诱导50% PBMC 和86% HLA-DR-CD4+T 细胞中HIV-1 的激活[52]。毛壳素+林立司他和毛壳素+prostratin 在重新激活感染者CD4+T 细胞中病毒方面比单用这2种化合物效力更高[52]。毛壳素细胞毒性较小,其与曲古抑菌素A和林立司他有协同作用,机制可能是通过染色体重构重新激活HIV-1基因的表达[53]。
2.5.2 BlX-01294
BIX-01294 可特异性抑制组蛋白甲基转移酶G9a 对H3K9的二甲基化(H3K9me2),对与其密切相关的胰高血糖素样肽酶(主要是H3K9me3)的抑制作用较弱,IC50分别为1.7 和38 μmol·L-1。研究表明,G9a C 端的SET 结构域是G9a 介导的H3K9甲基化所必需,同时也是抑制HIV-1 基因表达所必需[54]。HIV-1 基因通过招募G9a,导致H3K9me2 进而诱导基因沉默。随后H3K9me2 被包含异染色质蛋白1 和HDAC 的异染色质蛋白复合物识别,进而招募SUV39H,将沉默的常染色质转变为异染色质[54]。BIX-01294 抑制G9a,降低H3K9me2,并在转录水平重新激活潜伏的HIV-1。BIX-01294 可激活HIV-1感染者和无法检测到病毒载量患者的静息CD4+T细胞中HIV-1基因的表达[52]。
2.6 TLR激活剂
TLR 是一种模式识别受体(pattern recognition receptors,PRR)。PRR 主要是由免疫系统细胞表达的免疫受体,其功能是识别微生物特定分子结构即病原体相关分子模式(pathogen associated molecular pattern,PAMP)。TLR 为Ⅰ型跨膜蛋白,其胞外段为富含亮氨酸的重复序列,参与配体识别;胞内段含有保守的Toll 样/白细胞介素1(interleukin 1,IL-1)受体结构域,负责信号转导。TLR1,2,4,5,6 和10 定位于细胞表面,识别细菌和真菌的细胞壁成分。TLR3,7,8 和9 存在于细胞内,主要识别来自细菌和病毒的核酸。TLR 激活剂既是免疫激活剂,也是HIV 潜伏激活剂。TLR7 激活剂可诱导CK 的分泌,并使CD4+T 细胞中潜伏的HIV-1 重新激活。TLR9 激活剂MGN1703 可激活潜伏的HIV-1并增强抗病毒免疫反应[58]。TLR激活剂可通过上调CK 水平,增强免疫系统抗病毒的能力。目前大多数研究都集中在TLR7 和TLR9 激活剂。除能重新激活潜伏的HIV-1 外,TLR 通过同源二聚体或异源二聚体识别其同源配体,在巨噬细胞、PDC 和上皮细胞表面表达,介导对外来病原体的识别和应答。
2.6.1 维沙莫德(vesatolimod)
维沙莫德是一种可口服TLR7 激活剂,又称GS-9620,可直接激活PDC 和B 淋巴细胞,产生CK,诱导免疫激活。GS-9620 通过依赖于Ⅰ型干扰素的机制诱导接受HAART 患者细胞中的HIV-1重新激活,且增强抗体介导的免疫反应,最终提高对HIV-1 感染细胞的杀伤力[55]。研究表明,TLR2/7双重激活剂既能增强对潜伏病毒的再激活能力,又能增强免疫反应。2 类激活剂各自触发不同的途径,TLR2 激活剂Pam2CSK4 通过诱导记忆T 细胞中NF-κB 活化来重新激活HIV-1 基因表达,GS-9620 诱导单核细胞和PDC 分泌肿瘤坏死因子α,从而增强免疫反应[57]。另有研究表明,靶向HIV-1的广谱中和抗体PGT121 和GS-9620 联用能够延缓HIV-1 在停止服用HAART 药物的猴体内的再复制[56]。
2.6.2 来非莫德(lefitolimod)
来非莫德是一种TLR9 激活剂,又称MGN-1703,是一个含有非CG 甲基化的小DNA 分子。TLR9 主要表达于B 细胞和PDC。MGN-1703 可促进B 细胞分化,激活PDC、NK 细胞和T 细胞产生的干扰素,在淋巴结中发挥调节作用[59]。MGN-1703在CD4+T 细胞中促进HIV-1基因的转录,诱导强大的抗病毒天然免疫反应,并增强NK 细胞介导的抗病毒感染作用[58]。目前临床试验结果表明,MGN-1703耐受性良好,不良反应主要是1级或2级。
2.7 CK
CK 是免疫原、丝裂原或其他刺激剂诱导多种细胞产生的低相对分子质量可溶性蛋白质,具有调节固有免疫和适应性免疫、血细胞生成、细胞生长以及损伤组织修复等多种功能。CK 可被分为IL、干扰素、肿瘤坏死因子、趋化因子和生长因子等。CK 与细胞表面相应的受体结合后,可引发复杂的细胞内分子相互作用,最终导致细胞基因表达的改变。HIV-1 LTR 介导的转录受到一些CK 的调控。CK影响HIV-1基因转录的能力因细胞类型而异,并依赖于其他CK的存在[78]。
2.7.1 lL-7
IL-7 是趋化因子家族成员之一,是一种具有广泛免疫效应的多功能CK,其靶细胞主要为淋巴细胞,对来自人或小鼠骨髓的B祖细胞、胸腺细胞和外周T 细胞等均有促生长作用。IL-7 的受体是一个异源二聚体,由共有γ 链和IL-7受体特异性α 链组成。IL-7 通过与α 链结合,促使γ 链发生磷酸化,进而激活JAK/STAT 信号通路促进潜伏病毒的重新激活,同时IL-7 可通过促进外周CD4+T 细胞和CD8+T细胞的稳态增殖来恢复免疫功能,在病毒感染治疗时增强抗原特异性的T 细胞反应。目前IL-7 作为免疫激活剂已经进入临床研究[60],由于其对病毒的重新激活能力,未来可作为一种极具潜力的HIV-1 LRA。
2.7.2 N-803
N-803(ALT-803),是IL-15 超激动剂,是由突变的IL-15、IL-15受体α链和IgG1的Fc段融合而成的复合物。研究发现,在猴免疫缺陷病毒(simian immunodeficiency virus,SIV)感染的猕猴和HIV-1感染的人源化小鼠中,N-803 在CD8+T 淋巴细胞耗尽的情况下持续高强度诱导潜伏期逆转[61]。体外实验表明,N-803 能增强HIV-1 特异性CD8+T 细胞和NK 细胞介导的抗病毒免疫效应,从而靶向体内残留的病毒潜伏库[62]。同时N-803 激活的NK 细胞可抑制体内HIV-1 急性感染[63]。此外,在接受HAART 的猴-人类免疫缺陷病毒(simian-human immunodeficiency virus,SHIV)感染的猕猴中,皮下注射N-03 可促使NK 细胞和SHIV 特异性CD8+T细胞从外周血进入淋巴滤泡中[64]。
2.8 SMAC类似物AZD5582
SMAC 是存在于线粒体中调节细胞凋亡的蛋白质,通过阻碍凋亡抑制蛋白(inhibitor of apoptosis protein,CLAP),尤其是X 连锁凋亡抑制蛋白(X-linked inhibitor of apoptosis protein,XIAP)实现促凋亡作用。在CLAP1/2、肿瘤坏死因子受体相关蛋白组成的复合体中,CLAP1结构性降解NF-κB诱导激酶,从而阻止P100到P52的转化。SMAC类似物抑制CLAP,导致NF-κB诱导激酶积累和IKK磷酸化,以及随后P100到P52的加工,最后,P60/P52异源二聚体转移到细胞核,激活靶基因转录[79]。帕比司他和SMAC 类似物联合作用于接受HAART 患者的静息CD4+T细胞,可协同激活病毒潜伏库。
AZD5582 是一种新型小分子CLAP 抑制剂,可与CLAP1,CLAP2 和XIAP 的BIR3 区域有效结合,IC50分别为15,21 和15 nmol·L-1。HAART 联合AZD5582 处理可引起从BLT 小鼠和猕猴淋巴结及血浆中分离的T 细胞中HIV-1 RNA 和SIV RNA 水平显著增加,且毒副作用小。研究表明,AZD5582可以逆转静息CD4+T 细胞中HIV-1 和SIV 的潜伏状态[65]。
2.9 免疫检查点抑制剂
IC 分子是调节免疫反应的细胞表面受体。刺激性IC 分子可提供共刺激信号,增强免疫激活,而抑制性IC 分子会负调节免疫细胞功能,并在降低免疫效应和维持自身耐受性方面起重要作用[80]。在HIV-1感染期间,免疫细胞中抑制性IC分子上调,如程序性细胞死亡蛋白1(programmed cell death protein 1,PD-1)、细胞毒性T 淋巴细胞相关蛋白4(cytotoxic T-lymphocyte-associated protein 4,CTLA-4),会导致T细胞衰竭,其特征是免疫细胞的效应功能丧失并无法响应抗原而增殖[81]。抑制性IC 分子与HIV-1 潜伏库的建立和维持有关。HIV-1潜伏感染多发生在表达PD-1、T 细胞免疫球蛋白与黏蛋白结构域3、CTLA-4或B、T淋巴细胞弱化因子的细胞中[82]。此外,PD-1高表达的滤泡辅助性T细胞被证明富含复制能力强的HIV-1[83]。IC 分子如PD-1或CTLA-4主要通过干扰CD28信号通路阻断T 细胞受体传递信号。IC 抑制剂可促进T 细胞受体活化,激活下游信号转导通路,如酪氨酸激酶ZAP70、PI3K 和钙调神经磷酸酶等,进而激活调控HIV-1 基因转录的NF-κB,AP-1 和NFAT 等转录因子[84]。因此,IC 抑制剂可能是一种专门逆转HIV-1潜伏状态的LRA。
IC 抑制剂主要包括CTLA-4 抑制剂与PD-1/程序性细胞死亡-配体1(programmed cell deathligand 1,PD-L1)抑制剂,主要针对免疫T 细胞激活过程中的2 个关键IC 通路CTLA-4/B7-1/2 和PD-1/PD-L1通路。
2.9.1 纳武单抗(nivolumab)
纳武单抗是PD-1 抑制剂,可重新激活PD-1 过表达的HIV 特异性T细胞并提高其免疫应答。研究表明,在HIV-1感染的癌症病例中,纳武单抗成功增强了CD8+T 细胞增殖和分泌CK 的能力,扩大了PD-1低表达的T细胞亚群[66]。同样,在另一癌症病例中,纳武单抗治疗使得HIV-1 病毒潜伏库剧烈持续缩小,且CD8+T 细胞数量明显增加[67]。然而也有研究表明,将从接受HAART 患者体内分离的细胞进行体外培养,用PD-L1 抑制剂(BMS-936559)和纳武单抗联合治疗,不能持续促使PBMC 产生成熟病毒粒子[68]。在长期接受HAART 的SIV 感染的恒河猴中,双重阻断CTLA-4/PD-1通路可逆转潜伏病毒并缩小病毒潜伏库,但仍不足以完全控制病毒[69]。目前纳武单抗逆转潜伏HIV-1的作用机制尚不明了,需进一步研究。
2.9.2 伊匹单抗(ipilimumab)
伊匹单抗是CTLA-4 抑制剂。有研究表明,CD4+T细胞表面CTLA-4的高表达与HIV-1感染有关[70]。在接受HAART 的晚期恶性肿瘤病例中,单独使用PD-1 抑制剂纳武单抗对HIV-1 的潜伏状态或病毒潜伏库没有影响,但将纳武单抗和伊匹单抗联合使用可诱导相关未剪接的HIV-1 RNA 水平适度增加,并潜在消除了含有HIV-1 复制能力的细胞[71]。在3 例接受HAART 的肿瘤患者中发现,IC抑制剂伊匹单抗、纳武单抗和avelumab 可激活潜伏的HIV-1,增强HIV 特异性T 细胞功能,但个体差异较大[72]。在评估的剂量范围和时间范围内,未接受HAART 的患者使用伊匹单抗治疗是安全的且耐受性良好,且2剂以上更高剂量伊匹单抗(3 mg·kg-1)可极大促使T细胞激活和HIV-1基因表达[73]。
2.10 双硫仑(disulfiram)
双硫仑是特异性乙醛脱氢酶1 抑制剂,可通过对乙醇产生急性敏感性治疗慢性酗酒。双硫仑通过耗尽胞内磷酸酶和张力蛋白同源物,激活PI3K/Akt 信号通路,从而激活HIV-1 基因转录[74]。同时双硫仑治疗不会引起T 细胞的全局激活、促炎细胞因子的产生或激活标志物的表达。双硫仑与HDAC 抑制剂相似,其激活潜伏病毒的能力与缩减病毒潜伏库的能力无关。
3 结语
虽然HIV-1 LRA 的种类很多,但多数作用机制单一,不能有效激活病毒潜伏库,目前尚未发现能彻底激活HIV-1 病毒潜伏库的LRA,因此未来的研究应更倾向于不同作用机制药物的联用,这样不仅可有效提高激活效率,还可在一定程度上减轻单一药物的不良反应。在确定最佳LRA 组合和浓度等方面,既要做到协同效应最大化,又要确保彻底逆转潜伏病毒。研究表明,增加LRA 浓度,可增强逆转潜伏期的能力,但会降低LRA 间的协同作用[85]。除药物联用外,还可在给药方式和增加LRA 半衰期等方面进行优化,如采用靶向给药(如生物偶联)、纳米药物递送体系等毒副作用小、针对性强、且具有良好生物相容性的策略[14]。增加LRA 的半衰期可通过延长药物在体内的留存时间来实现,避免由于接触时间过短,而降低药物作用效率。但LRA 的使用也有很多限制,如某些LRA 会破坏内皮屏障的完整性[86];由于LRA 缺乏特异性,不同感染者有不同的激活情况,即病毒潜伏库的异质性和导致HIV-1潜伏机制的多样性,潜伏库的异质性在很大程度上导致LRA 的临床试验成功率低[87]。其他问题还包括:研究人员尚不清楚体外“效力”是否与体内“效力”有关,特别是消除潜伏感染细胞的能力;残留的病毒血症是否表示在接受HAART 后病毒的持续复制,还是病毒从激活的宿主中释放;LRA 干预前潜伏库的大小可能会影响潜伏期逆转[88]。此外,试验中使用的一些LRA 导致CD8+T 细胞功能障碍,并使潜伏感染细胞上T 细胞耗竭标志物上调,从而降低细胞免疫反应[89-90]。这些问题有待进一步深入研究。
