铅和镉单独或联合暴露致中枢神经毒性及机制研究进展
2023-08-01黎婵华罗京京彭健超黄诗卉谭翠奇姜岳明
黎婵华,罗京京,彭健超,黄诗卉,谭翠奇,姜岳明
(广西医科大学公共卫生学院毒理学系,广西 南宁 530021)
铅和镉在工业中应用广泛,其污染环境,并对人体健康产生严重的影响。铅和镉主要在有色金属的加工冶炼过程中产生,在水源、土壤、食物等环境介质中广泛存在。近年来发现,在香烟烟雾和电子烟中也有铅、镉污染,长期低水平铅、镉暴露可能会对神经系统造成严重的影响或损害[1-2]。皮质神经细胞同时铅和镉暴露可引起细胞数量减少。妊娠大鼠经饮水暴露于铅和镉,可使子代大鼠脑发育迟缓,皮质超微结构异常。铅主要蓄积在骨骼,镉以肾和肝蓄积为主,二者也可在大脑蓄积,引起神经系统毒性,其毒作用机制可能与氧化应激、体内必需微量元素分布紊乱、线粒体功能障碍、细胞凋亡和表观遗传失调等有关[3]。为此,本文就铅和镉单独或联合暴露导致的中枢神经毒性研究进展做一综述,为深入了解其毒作用机制及寻找相应防治对策提供理论依据。
1 铅暴露的中枢神经毒性及机制
1.1 铅暴露可能引起的神经系统疾病
1.1.1 阿尔茨海默病(Alzheimer disease,AD)
中枢神经系统是铅作用的靶器官之一,铅中毒可以导致儿童和成年人认知功能障碍、记忆缺陷和智力下降等AD 症状。根据2008 年美国健康营养调查研究(包括8080 名老年人),调整人口学资料和生活方式的混杂因素后,随着血铅水平的增加,AD死亡风险增加[4]。婴儿和胎儿时期铅暴露可能对成年期产生长期影响,与大脑功能异常、认知功能缺陷以及神经退行性疾病如AD等相关[5-7]。80只子代雄性SD 大鼠分成4 组,分别饮用含铅0,0.5‰,1‰和2‰的蒸馏水4 周,发现铅暴露浓度与皮质和海马区β 淀粉样蛋白42(amyloid β-protein 42,Aβ42)表达量和淀粉样斑块密度呈正相关,铅暴露增加皮质和海马区Aβ42的表达,以致淀粉样斑块沉积加重[8]。与成年期铅暴露比较,大鼠出生后用含铅0.1%的饮用水持续暴露至72周龄,海马tau蛋白磷酸化和Aβ 水平升高,并损伤其记忆和学习能力[9]。铅暴露随访研究显示,儿童出生后早期铅暴露可能与老年时期大脑形态结构改变和功能代谢的损伤有关[10]。
1.1.2 孤独症谱系障碍(autism spectrum disorders,ASD)
铅中毒与儿童的社会退缩、沟通障碍、记忆力下降和智商降低等症状有关[11]。据一项1∶1 病例对照研究显示,ASD 患儿血液和头发中铅含量明显增加,使用铅螯合剂2,3-二巯基丁二酸治疗后,其语言能力随着血铅含量的降低得到明显改善,提示铅毒性与ASD 的发病存在明显相关性。体内铅的蓄积可能是由于ASD 患儿代谢缺陷和排铅能力降低所致。因此,使用螯合剂降低体内铅水平可改善ASD 症状[12]。对2473 例7~8 岁ASD 患儿的随访研究发现,调整家庭因素(如父母教育背景、家庭收入和是否吸烟等)和儿童出生指标(胎龄和出生体重等)后发现,即使患者在7~8 岁时血铅浓度较低(几何均数为1.64 μg·dL-1),其在11~12 岁时的ASD 症状也与血铅浓度相关,提示需要进一步降低铅暴露水平[13]。对30 例ASD 患儿和30 例健康对照组儿童进行发铅浓度检测发现,ASD 患儿发铅浓度为(6.028±0.690)mg·kg-1,约为对照组〔(3.415±1.207)mg·kg-1〕的2 倍,提示儿童发铅浓度与ASD有关[14]。此外,妊娠期母亲的血铅水平与儿童患ASD风险存在明显相关性[15]。
1.1.3 其他疾病
1972-1973 年新西兰出生队列前瞻性研究显示,经过调整母亲、儿童智商和社会经济状况等因素后,儿童血铅水平每升高5000 μg·L-1,成人期智商水平降低1.61分,诊断性访谈量表(DIS-Ⅳ)[16]的评分增加1.34 分,提示儿童期血铅水平与成人期智商下降和罹患精神疾病风险增加有关[17-18]。1 项包括101 例帕金森病(Parkinson disease,PD)患者(病例组)和50例健康志愿者的病例对照研究发现,PD 患者累积铅暴露浓度(以胫骨和髌骨铅浓度评估铅蓄积暴露浓度)高于对照组,且随铅暴露浓度的升高,PD 患者认知能力下降,提示铅暴露与PD发生风险显著相关,铅暴露可能使PD 患者的认知损害加重[19]。40 只听力正常的成年豚鼠ig 给予醋酸铅2 mmol·L-10,15,30,60 和90 d 后,随着染铅时间延长,听觉脑干反应Ⅰ波潜伏期、Ⅲ波潜伏期和Ⅰ~Ⅲ波间隔均延迟,60 d 后螺旋神经节神经元数量逐渐减少,自噬相关蛋白5、自噬相关蛋白6 和微管相关蛋白1 轻链3B 在脑干的表达增加,提示铅暴露可使脑干听觉功能下降,以致损伤听力。推测听觉神经传导通路(包括螺旋神经节神经元和脑干)是铅中毒的主要靶点,自噬是铅对听觉神经系统的毒性作用机制之一[20]。将5 周龄健康雄性小鼠分为染毒组和对照组,分别ip 给予醋酸铅5,7 和12 mg·kg-1或等量生理盐水2周以建立慢性铅中毒小鼠模型后,与对照组比较,染毒组小鼠出现抑郁样症状,额叶5-羟色胺(5-hydroxytryptamin,5-HT)含量随染铅浓度增加而逐渐减少,推测铅中毒后出现抑郁症状可能与额叶5-HT 含量失衡有关[21]。雌性SD 大鼠在人工交配成功后开始醋酸铅5 mg·L-1染毒,大鼠分娩后继续染铅至其子鼠断奶后5周龄,与对照组比较,染毒组子鼠血液和海马铅含量明显增加,并出现注意缺陷多动障碍(attention deficit hyperactivity disorder,ADHD),说明血液和海马中的铅水平与ADHD的发生具有相关性[22]。
1.2 铅暴露致神经损伤机制
铅进入血液后,可替代Ca2+穿过血脑屏障和胎盘屏障进入神经细胞,在大脑脉络丛蓄积,造成铅中毒[23]。铅可干扰Ca2+信号传递,取代Ca2+蛋白质结合位点,调节阳离子通道的活性,增加细胞内游离Ca2+浓度,导致细胞内线粒体和内质网Ca2+浓度增加,上调1,4,5-三磷酸肌醇受体3和雷诺丁受体的表达和激活,影响细胞外信号调节激酶(extracellular signal-regulated kinase,ERK)/Bcl-2蛋白通路,以致细胞凋亡。铅进入细胞内部后,促进内质网Ca2+释放,导致钙调蛋白依赖性蛋白激酶Ⅱ(calmodulin-dependent protein kinasesⅡ,CaMKⅡ)的磷酸化,引起转录因子环磷腺苷效应元件结合蛋白表达水平下降,提示铅通过抑制Ca2+/血清钙调蛋白/CaMKⅡ/环磷腺苷效应元件结合蛋白信号通路,下调下游分子早期生长反应因子1 和碱性成纤维细胞生长因子的表达水平,减弱神经细胞再生功能,从而损伤长期记忆与认知功能。铅能够抑制沉默信息调节因子1和腺苷酸活化蛋白激酶信号转导途径,影响细胞信号转导功能,干扰正常的突触信号传递,促进细胞凋亡、钙平衡失调、自噬和能量代谢平衡紊乱。铅暴露增强Bax 和P53 蛋白表达,Bax/Bcl-2 比例失衡,诱导线粒体障碍,促进活性氧(reactive oxygen species,ROS)释放和脂质过氧化物(lipid peroxides,LPO)生成,导致细胞凋亡。铅离子通过Toll 样受体4/髓样分化因子88/NF-κB信号通路导致小胶质细胞和星形胶质细胞增多,促进炎症介质的表达,增加炎症因子的释放,以致产生神经炎症。此外,铅暴露干扰核转录因子红系2相关因子2(nuclear factor erythroid 2-related factor 2,Nrf2)/血红素加氧酶1/磷脂酰肌醇3 激酶(phosphoinositide 3-kinase,PI3K)/糖原合成酶激酶3β信号通路,阻断蛋白激酶B(protein kinase B,Akt)/哺乳动物雷帕霉素靶蛋白(mammalian target of Rapamycin,mTOR)信号通路,Akt 和mTOR 蛋白水平降低,减少氧化应激分子超氧化物歧化酶(superoxide dismutase,SOD)、过氧化氢酶(catalase,CAT)和谷胱甘肽的表达水平,提示铅暴露可以负向调节PI3K/Akt/mTOR 信号通路,诱导氧化应激[24]。据NOD 样受体热蛋白结构域相关蛋白3(NOD-like receptor pyrin domain-associated protein 3,NLRP3)炎症小体敲除小鼠体内实验研究,铅暴露使海马区NLRP3炎症小体表达增强,并促进胱天蛋白酶1 在信使RNA 和蛋白质水平上的激活,表明NLRP3炎症小体参与神经炎症的发生,导致海马区神经元损伤和小胶质细胞活化[25]。通过采用幼虫喂养技术对过度表达SOD和CAT 以及敲除SOD 和CAT的黑腹果蝇染铅0.2~0.8 mmol·L-1,发现敲除SOD 和CAT 的黑腹果蝇大脑铅负荷增大,引起DNA 损伤、细胞凋亡、氧化应激成分与空泡化增加,以致神经元功能障碍和自噬失调。铅暴露可引起大脑神经细胞氧化应激和DNA 损伤等[26]。铅还可通过多种机制损害神经递质受体功能,如铅和锌竞争性结合N-甲基-D-天冬氨酸受体(N-methyl-Daspartate receptor,NMDAR)GluN2 亚基的锌结合位点,从而改变GluN2 亚基的基因和蛋白质表达,进而抑制海马NMDAR受体依赖性长时程增强和突触可塑性。此外,还能通过取代特异性蛋白1 中的锌改变NMDAR 结构和基因表达[27],而NMDAR功能障碍与AD的发生发展有关[28]。铅暴露致神经损伤可能的作用机制如图1所示。
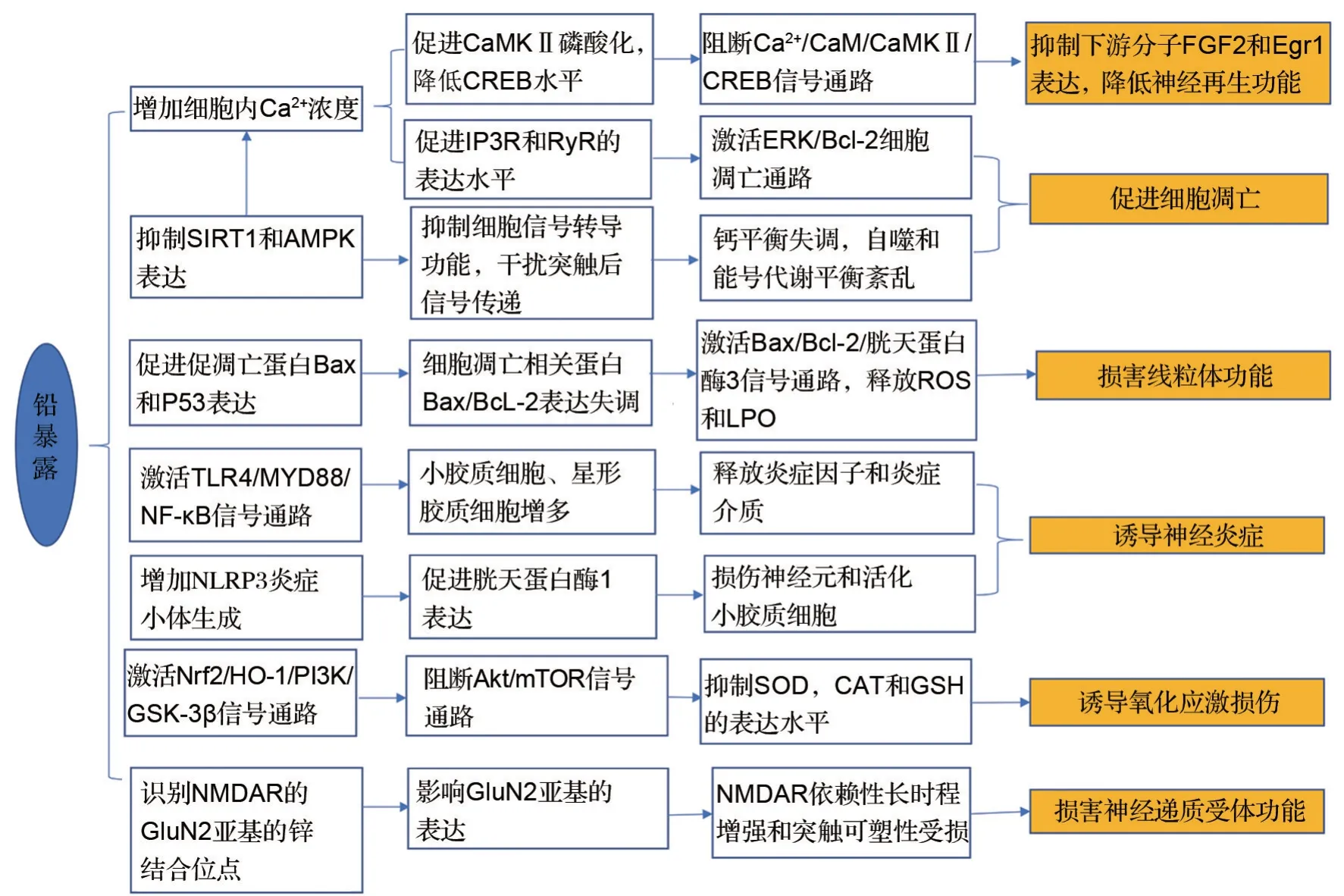
图1 铅暴露致神经损伤可能的作用机制. SIRT1:沉默信息调节因子1;AMPK:腺苷酸活化蛋白激酶;NMDAR:N-甲基-D-天冬氨酸受体;CaM:血清钙调蛋白;CaMKⅡ:钙调蛋白依赖性蛋白激酶Ⅱ;CREB:环磷腺苷效应元件结合蛋白;IP3R:1,4,5-三磷酸肌醇受体3;RyR:雷诺丁受体;Akt:蛋白激酶B;mTOR:哺乳动物雷帕霉素靶蛋白;ERK:细胞外信号调节蛋白激酶;ROS:活性氧;LPO:脂质过氧化物;Nrf2:核转录因子红系2相关因子2;HO-1:血红素加氧酶1;PI3K:磷脂酰肌醇3激酶;GSK-3β:糖原合酶激酶3β;TLR4:Toll样受体4;MYD88:髓样分化因子88;NLRP3:NOD 样受体热蛋白结构域相关蛋白3;SOD:超氧化物歧化酶;CAT:过氧化氢酶;GSH:谷胱甘肽;FGF2:碱性成纤维细胞生长因子;Egr1:早期生长反应因子1.
2 镉暴露的中枢神经毒性及机制
2.1 镉暴露可能引起的神经系统疾病
镉暴露与多种神经系统疾病的发生存在关联,如AD,PD,ASD 和ADHD,这些神经系统疾病的发生风险随着镉暴露浓度的增加而增加,其流行病学研究进展见表1。

表1 镉暴露可能引起的神经系统疾病流行病学研究
2.2 动物实验研究
将人类载脂蛋白E3(apolipoprotein E 3,ApoE3)或ApoE4基因导入小鼠体内,当该小鼠经饮用水暴露于氯化镉(0.6 mg·L-1)14 周后,出现空间工作记忆缺陷,导入ApoE4基因的小鼠中毒症状更严重,提示ApoE4 和环境镉暴露可能存在相互作用,使认知障碍加重,以致小鼠神经元退行性病变[38]。大鼠持续4 周每周累积染镉50 mg·kg-1,可出现焦虑、抑郁样行为和记忆功能受损[39]。每天ip给予大鼠氯化镉1 mg·kg-1,持续3 个月后,受试大鼠在Y 迷宫实验中自发性交替反应率减低,推测镉暴露可损害大鼠空间记忆功能[40]。
2.3 镉暴露致神经损伤机制
镉能穿过血脑屏障,在脉络丛的蓄积量比大脑皮质和脑脊液高,并损害其结构[41]。镉进入大脑神经元,会抑制SOD、谷胱甘肽过氧化物酶和CAT 活性,促进LPO 生成[42]。镉激活突触后膜瞬态电压感受器阳离子通道亚家族V 成员1,促进线粒体产生ROS,导致细胞生成LPO,神经递质释放受阻[43]。镉可以激活丝裂原活化蛋白激酶/mTOR 和肿瘤坏死因子受体相关因子6/NF-κB抑制蛋白α/NF-κB诱导细胞凋亡和自噬,并通过介导腺苷酸活化蛋白激酶/过氧化物酶体增殖物激活受体γ 共激活因子1α/Nrf1/2信号通路,促进胱天蛋白酶8和c-Jun N端激酶的表达,诱导线粒体损坏和凋亡。镉上调大鼠皮质神经元胱天蛋白酶8、胱天蛋白酶3、聚腺苷二磷酸核糖聚合酶1 和凋亡信号调节激酶1 的表达,影响线粒体Bcl-2 和Bax 表达,导致自噬功能缺失和细胞凋亡[44-46]。镉可降低大鼠皮质和海马Bcl-2/Bax 比值,导致细胞凋亡,胱天蛋白酶3/7 的表达与镉浓度呈剂量-效应关系,推测镉诱导细胞凋亡可能通过激活胱天蛋白酶3/7所致[47]。镉通过抑制线粒体ROS 依赖性的Akt/糖原合成酶激酶3β/β 连环蛋白信号通路,干扰小鼠神经干/祖细胞的生长、增殖和存活,使神经胶质纤维酸性蛋白(星形胶质细胞活化标志物)水平升高,并诱导白细胞介素6 的释放,抑制大脑纤毛基因表达[48-49]。镉暴露促进小鼠齿状回颗粒下区神经干/祖细胞分化,引起海马神经元数量减少,以致损害学习记忆能力[50]。镉诱导SH-SY5Y 细胞产生金属硫蛋白(metallothionein,MT),MT 在室管膜细胞和脉络丛上皮细胞积累,镉离子和MT 结合,进而干扰必需金属元素对矿物质的吸收[51]。染镉导致斑马鱼的运动能力和对环境信号的反应力受到影响,细胞周期停滞和凋亡,激活小胶质细胞并引起神经炎症[52]。镉暴露能够影响糖酵解、色氨酸、脂肪和酪氨酸等代谢通路,染镉剂量越高,对代谢通路的影响越严重,以致细胞膜受损,能量供应和氧化还原失衡,最终导致细胞凋亡[53]。染镉可降低大鼠海马神经元存活率,影响学习与记忆相关受体和蛋白质表达水平,如降低NMDAR 的NR2A 亚基和突触后致密蛋白9、脑源性神经营养因子、酪氨酸蛋白激酶受体B和ERK1/2的表达水平[54]。给亲代斑马鱼(F0 代)染氯化镉0.01 μmol·L-160 d 后,子代斑马鱼(F1 代)的运动能力明显下降,如游泳速度减慢和游泳距离缩短,神经递质如多巴胺、5-HT 和乙酰胆碱表达水平降低,神经发育和神经递质代谢相关基因表达也发生改变,导致F1代斑马鱼发育性神经毒性[55]。基底前脑胆碱能神经元可调节认知功能,甲状腺激素可维持这些神经元的功能,镉可破坏胆碱能神经元,增加促甲状腺激素的释放,使T3和T4水平下降,进而破坏神经系统镉拮抗毒蕈碱受体,降低乙酰胆碱水平,以致胆碱能神经元死亡,大脑认知功能受损[56]。此外,镉导致炎症小体NLRP3 大量产生,通过白细胞介素1β/NF-κB 抑制蛋白α/NF-κB/NLRP3 的正反馈回路诱导神经炎症[57],镉暴露致神经损伤可能的作用机制如图2所示。
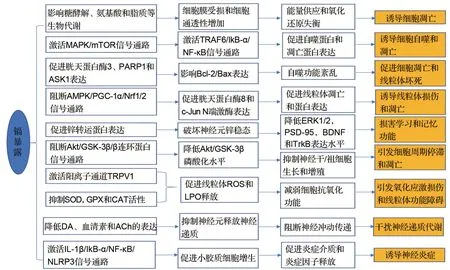
图2 镉暴露致神经损伤的可能的作用机制. MAPK:丝裂原活化蛋白激酶;PARP1:聚腺苷二磷酸核糖聚合酶1;ASK1:凋亡信号调节激酶1;PGC-1α:过氧化物酶体增殖物激活受体γ 共激活因子1α;PSD-95:突触后致密蛋白95;BDNF:脑源性神经营养因子;TrkB:酪氨酸蛋白激酶受体;TRPV1:瞬态电压感受器阳离子通道亚家族Ⅴ成员1;IkB-α:NF-κB 抑制蛋白α;DA:多巴胺;ACh:乙酰胆碱;ROS:活性氧;TRAF6:肿瘤坏死因子受体相关因子6;IL-1β:白细胞介素1β;JNK:c-Jun N端激酶;GFAP:胶质纤维酸性蛋白.
3 铅镉联合暴露的中枢神经毒性及机制
职业性铅镉暴露多见于电池回收和焊接过程,如有色金属冶炼、蓄电池制造业和汽车厂油漆工人血铅、镉的含量明显高于非暴露者,环境铅镉污染常常存在,铅和镉联合污染常存在于高速公路的尾气、工业废水和空气气溶胶中[58]。其暴露形式不仅存在铅和镉的单独暴露,还有铅镉联合暴露,损害神经系统[59]。香烟烟雾也是铅和镉的重要来源,这些金属颗粒会沉积在环境中,如家具、地板和墙壁等,污染生活环境。
3.1 与铅镉联合暴露有关的神经系统疾病
在一项孕晚期铅和镉暴露对婴儿神经心理发育和动作能力(粗大和精细动作)的纵向调查中,在调整母亲教育水平、经济收入、年龄和喂养状况后,使用贝利婴幼儿发展量表评估婴儿的认知发育情况,发现当镉浓度>1.51 μg·L-1时,孕晚期铅镉联合暴露可能增加婴儿认知发育障碍的风险[60]。儿童经水、土壤和食物等途径暴露于铅镉后,其血液中铅和镉浓度随年龄增长有所增加,污染区儿童体内体格发育相关生长激素(生长激素、胰岛素生长因子1 和生长激素结合蛋白等)和甲状腺激素水平降低,导致儿童智力和体格发育障碍[61]。妊娠期母鼠持续ig给予铅300 mg·L-1和镉10 mg·L-1至仔鼠断奶。与对照组比较,铅镉联合暴露组仔鼠的焦虑指数升高,海马多巴胺含量减少,5-HT含量升高,提示铅镉联合暴露可能会累及大鼠中枢单胺能神经系统[62]。妊娠期SD 大鼠暴露于铅300 mg·L-1和镉10 mg·L-121 d,新生仔鼠海马铅和镉水平明显升高,水迷宫实验逃避潜伏期延长,进入盲端次数增多,提示铅镉联合暴露可能使子代大鼠认知能力降低[63]。
3.2 铅镉联合暴露致神经损伤的可能机制
铅和镉的化学特征和在体内的吸收转运机制相似,穿过血脑屏障进入中枢神经系统后消耗体内的抗氧化物酶,如酸性磷酸酶、CAT和ATP 酶等,从而破坏体内抗氧化防御系统,增强对中枢神经系统的氧化应激作用[64]。铅和镉联合暴露可导致浦肯野纤维细胞层出现多层结构,大脑皮质神经元形态发生改变,脑干和小脑星形胶质细胞活化增生,以致发生神经炎症[65]。铅镉联合暴露(铅10 μmol·L-1+镉2.5 μmol·L-1)使SH-SY5Y 细胞存活率降低,细胞内ROS 和细胞核内Nrf2 水平升高,提示铅镉联合暴露使Nrf2 从细胞质转移到细胞核内。此外,Nrf2下游调控分子HO-1蛋白的表达水平也有所提高,提示铅镉联合暴露可诱导SH-SY5Y 细胞氧化应激,产生细胞毒性[66]。同时铅镉联合暴露使PC12细胞的突起长度变短,分支数量减少,对细胞突起产生抑制作用,当铅浓度为2.5~20 μmol·L-1时,抑制作用呈浓度依赖性,且铅镉联合暴露产生的细胞毒性明显高于铅或镉单独暴露的毒性[67]。SD大鼠同时ig 给予镉50 mg·L-1和铅300 mg·L-1,持续12 周,暴露组大鼠皮质凋亡相关因子配体、蛋白激酶R 样内质网激酶和转录激活因子等蛋白表达水平明显比铅或镉单独暴露组高,提示铅和镉单独或联合暴露可能会激活Fas/FasL 和线粒体凋亡途径,诱导内质网应激,损害神经细胞。与铅或镉单独暴露组比较,铅镉联合暴露对大脑皮质损伤更严重[68]。SD 大鼠铅镉单独或联合暴露后,其海马组蛋白去乙酰化酶2(histone deacetylase 2,HDAC2)表达量均有所升高,铅镉联合暴露组HDAC2 表达水平高于铅或镉单独暴露组,表明铅联合暴露可能经HDAC2 的表观遗传调控机制损害神经系统[67]。Wistar 大鼠单独或联合ig 给予含铅或镉的饮用水后,大鼠脑氧化应激标志物硫代巴比妥酸反应物和丙二醛水平均升高,大鼠脑氧化-还原状态失衡,提示铅镉联合暴露可经氧化应激途径损伤神经系统[69]。
4 结语
综上,铅镉单独或联合暴露与神经退行性疾病的发生发展有关,对其神经毒作用机制的研究可能为中毒防治干预提供新思路。但是,铅和镉联合暴露的体内外神经毒性实验和人群调查资料尚十分有限,相关毒作用机制研究也不够深入。尽管环境与职业铅镉暴露水平、人群体内重金属安全限值不断下降,美国疾病预防控制中心也建议不高于50 μg·L-1作为血铅暴露的安全限值。然而,低水平铅和镉暴露对人群也可能产生有害的影响或潜在毒作用,目前尚无铅镉联合暴露的安全限值,仍需深入探究铅镉联合暴露的中枢神经毒性机制,阐明其对人体健康的影响,为制定有效的铅镉中毒防治措施提供科学依据。
