基于Adaptive Lasso的两阶段全基因组关联分析方法
2023-07-20杨文宇吴成秀肖英杰严建兵
杨文宇 吴成秀 肖英杰,3,* 严建兵,3
基于Adaptive Lasso的两阶段全基因组关联分析方法
杨文宇1,2吴成秀1肖英杰1,3,*严建兵1,3
1作物遗传改良全国重点实验室, 湖北武汉 430070;2华中农业大学理学院, 湖北武汉 430070;3湖北洪山实验室, 湖北武汉 430070
作为进行全基因组关联分析的主流方法, 混合线性模型类方法得到了广泛的应用。但是, 现有方法仍存在检测功效不高的问题。本文提出一种基于Adaptive Lasso的2阶段全基因组关联分析方法(two-stage Adaptive Lasso-based genome-wide association analysis, ALGWAS), 该方法在第1阶段通过变量选择方法Adaptive Lasso筛选出与目标性状相关联的单核苷酸多态性位点(single nucleotide polymorphism, SNP), 第2阶段将第1阶段筛选出的SNP作为协变量放入线性模型中进行全基因组扫描。在模拟实验中, ALGWAS方法与3种常用的全基因组关联分析方法fastGWA、GEMMA和EMMAX相比具有最高的检测功效, 同时具有较低的错误发现率(false discovery rate, FDR)。将以上4种方法应用到包含1341份材料的玉米CUBIC (Complete-diallel plus Unbalanced Breeding-like Inter-Cross)群体的全基因组关联分析中, ALGWAS方法可检测到与开花期相关基因、和, 与株高相关基因和, 与产量相关基因、和等, 而其他3种常用的全基因组关联分析方法检测功效较低。本研究提出了一种非混合线性模型类的全基因组关联分析方法, 对解析微效多基因决定的复杂遗传性状具有更高的检测效率, 为基因挖掘提供了新的途径。
玉米; 全基因组关联分析; 变量选择; Adaptive Lasso
全基因组关联分析(Genome-Wide Association Study, GWAS)是在全基因组水平上分析高密度的SNP与性状相关性的分析, 从而发现影响复杂性状的基因变异的一种统计方法。遗传学家最先使用的是简单易算的线性模型(Linear Model, LM), 但该模型没有考虑群体结构的影响, 会挖掘出很多基因位点与复杂性状的假阳性关联。在一般线性模型中控制群体结构效应后, 假阳性检测大大降低。此外, 遗传学家发现复杂的亲缘关系也会带来假阳性的关联结果, 因此Zhang等[1]和Yu等[2]提出了混合线性模型。混合线性模型能同时控制群体结构和亲缘关系的影响, 降低了假阳性率。此后, 很多研究者致力于改善混合线性模型。Kang 等[3]2008年提出有效的混合线性模型(Efficient Mixed-Model Association, EMMA)。EMMA是一种被广泛使用的精确方法, 它将求解混合线性模型时涉及的优化问题转化成了一维的优化问题, 提高了计算效率, 并通过谱分解方法避免了每次迭代计算似然函数时的大量矩阵相乘和求逆运算, 进一步提高了计算效率。但是EMMA难以处理由数千个个体组成的数据集, 为了解决这个问题, Kang等[4]2010年在EMMA的基础上提出了EMMAX (EMMA eXpedited)。EMMAX将EMMA扫描每个位点时均需估计的多基因方差与残差方差之比用无效应模型中得到的比值近似, 大幅减少了计算量。同年, Zhang等[5]在混合线性模型的基础上提出了压缩的混合线性模型(Compressed MLM, CMLM)和P3D (Population Parameters Previously Determined)方法。CMLM采用聚类方法将群体进行分组, 减少了有效样本数量。P3D通过固定多基因方差与残差方差的比值, 减少了全基因组扫描时需要估计的参数数目, 提升了计算效率。2012年Zhou等[6]提出一种高效的精确方法, 全基因组高效混合线性模型(Genome- wide Efficient Mixed-Model Association, GEMMA)。GEMMA大约比EMMA快倍(为样本数目), 它的出现使得处理大样本数据集时采用精确全基因组关联分析方法变得可行。近年来, 混合线性模型类方法得到了广泛的应用[7-13], 与之相关的快速算法也陆续被提出, 如Fast-LMM[14]、Fast-LMM-Select[15]和BOLT-LMM[16]等。2019年Jiang等[17]针对大规模数据分析, 开发了一种基于混合线性模型的新方法fastGWA, 它通过将亲缘关系矩阵中较小系数替换成0值, 增加矩阵稀疏性, 提高了模型功效和运算速度, 并用模拟实验证明了fastGWA的可靠性和鲁棒性。
在过去的几十年, GWAS在人类、动物和植物中识别了成千上万的相关基因座, 为疾病诊断和动植物育种提供了帮助。但是, GWAS识别出的基因座只能解释很小的一部分表型变异, 这种现象被称为“消失的遗传力”[18]。例如, GWAS识别到了约50个与人类身高相关的基因座, 但是他们仅能解释5%的身高变异[19]。Yang等2010年指出遗传力并没有消失, 而是基因组中存在大量的微效位点GWAS检测不到[20]。这说明长期以来复杂性状GWAS一直都存在检测功效不足的问题。为了提高GWAS的检测功效, 主要有以下3个方面的探索: (1) 增加标记的类型, Song等[21]采用InDel (short insertion/deletion)作为标记进行 GWAS分析, 发现使用SNP进行GWAS检测不到的基因; (2) 采用多变量模型, Zhang等[22]通过模拟实验和真实数据验证了多位点模型MrMLM的优越性; (3) 采用非参数模型, Yang等[23]提出A-D test方法, 对不服从正态分布的表型可提高GWAS的检测功效。本研究在参数模型的范畴下, 为了提高GWAS的检测功效提出一种基于Adaptive Lasso的2阶段全基因组关联分析方法(ALGWAS), 该方法先通过Adaptive Lasso筛选出与目标性状相关联的SNP, 再将筛选出的SNP作为协变量放入一般线性模型中进行全基因组扫描。本研究选用包含1341份材料的玉米CUBIC群体的基因型和模拟的表型, 采用2种模拟方法进行数值实验, 并与3种常用的全基因组关联分析方法fastGWA、GEMMA和EMMAX进行对比。试验结果显示ALGWAS具有最高的检测功效且具有较低的错误发现率。
本文使用以上4种方法对玉米CUBIC群体的开花期、株高和产量数据进行全基因组关联分析, 发现ALGWAS方法可检测到与开花期相关的已知基因、和, 与株高相关的已知基因和, 与产量相关的已知基因、和等, 而其他3种常用的全基因组关联分析方法只能检测到少量已知基因。
1 材料与方法
1.1 试验材料的基因型和表型
本研究所用的1341份材料来源于玉米CUBIC群体[24]。该群体通过以“黄改系”为核心的24个优良玉米自交系作为亲本, 采用一代不完全的双列杂交和6代的随机交配, 再进行6代的连续自交得到。利用第2代测序技术对CUBIC群体的1341个后代自交系进行低覆盖度的测序(~1X), 选择最小等位基因频率大于0.02, 获得11,800,000高质量的SNP, 本文从中随机挑选标记60,000个。在全国选取5个典型玉米种植生态区种植CUBIC群体, 进行大规模的田间表型试验。对每份材料调查抽雄期(days to tasseling)、株高(plant height)和穗重(ear weight)性状。本研究利用的基因型和表型性状数据来自Liu等[24]已发表文章。
1.2 模拟试验方案
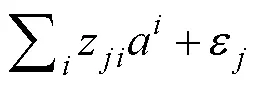
1.3 ALGWAS方法
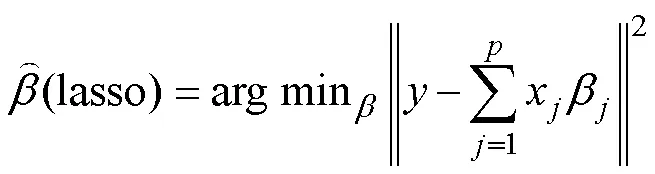
1.3.2 ALGWAS方法的第2阶段 假设第1阶段由Adaptive Lasso筛选出与性状相关的SNP集合为B, 第2阶段全基因组扫描到第个SNP, 定义ALGWAS方法第2阶段的模型为:
=+zγ+, (1)
这里是×1表型向量,z是×1基因型向量,γ是第个SNP效应,=[1, …,x+1]是×(+1)设计矩阵,x+1=(1, …, 1),=(1, …,b+1)是(+1)×1系数向量,b+1为模型(1)的截距,为集合B中选出进入模型(1) SNP的个数(<||),(0,2)。规定扫描窗口大小为10 Mb, 即第个SNP左右5 Mb以外的集合B中的SNP, 作为检测第个SNP的协变量进入模型(1)。ALGWAS 方法的R语言程序可从github (https://github.com/yangwenyurain/ALGWAS. git)下载。
1.4 常用GWAS方法
1.4.1 线性模型(LM)方法 线性模型为:=+Zγ+, 这里是×1表型向量,是截距,Z是×1基因型向量, γ是第个SNP的效应,(0,2)。
1.4.2 混合线性模型方法 混合线性模型为:=++,是×1表型向量,是固定效应对应的×设计矩阵,是×1代表固定效应的系数向量,是随机效应对应的×设计矩阵, 多基因效应(0,2),为亲缘关系矩阵, 残差效应向量(0,2),为单位矩阵,2和2分别为估计的遗传方差和残差方差。本研究利用EMMAX[4]、GEMMA[6]和fastGWA[17]3种常用的混合线性模型进行模拟数据和真实数据的GWAS分析。
2 结果与分析
2.1 模拟试验方案1: 从头模拟表型
利用CUBIC群体基因型数据, 定义20个和50个QTN, 狭义遗传力为0.5和0.8, 共4个模拟组合, 随机重复50次后, 共得到200个模拟表型。使用LM、EMMAX、GEMMA、fastGWA和ALGWAS分别对其进行全基因组关联分析, 得到的平均结果见表1。从表1可以看出, ALGWAS与EMMAX、GEMMA和fastGWA相比具有最高的平均检测功效和较低的错误发现率, 进一步可以看出ALGWAS检测功效高的原因在于ALGWAS对于低效应的QTN平均检测功效比较高。当QTN数目为20, 遗传力为0.8时, ALGWAS的平均检测功效为0.802, fastGWA的检测功效为0.457, ALGWAS对于低效应QTN的平均检测功效为0.48, 比fastGWA的平均检测功效0.04高12倍。
2.2 模拟实验方案2: 基于真实性状遗传结构的表型模拟
在CUBIC群体观察到的表型抽雄期、株高和穗重上分别随机选择标记, 添加1个QTN效应, QTN的效应设置为表型标准差的0.1倍至0.5倍, 重复50次后, 共得到1350个模拟表型。使用EMMAX、GEMMA、fastGWA和ALGWAS分别对其进行全基因组关联分析, 得到的平均结果如图1。从图1可以看出在不同表型上添加QTN效应, ALGWAS均具有最高的平均检测功效, 尤其是添加小效应QTN时, ALGWAS的优势更明显, 例如在穗重表型上添加表型标准差0.1倍的QTN效应时, EMMAX、GEMMA和fastGWA的平均检测功效均为0, 而ALGWAS的检测功效为0.12。
2.3 真实数据结果
考虑CUBIC群体观察到的表型抽雄期、株高和穗重, 使用EMMAX、GEMMA、fastGWA和ALGWAS分别对其进行全基因组关联分析(图2~图4)。可以看出EMMAX、GEMMA和fastGWA方法检测到的QTL, ALGWAS均可检测到, 并且ALGWAS还可检测到更多的QTL, 这说明了ALGWAS有更高检测功效。对于抽雄期, ALGWAS方法可检测到与开花期相关的基因、和[24,29], 而GEMMA和fastGWA只能检测到基因和, EMMAX仅能检测到基因。对于株高, ALGWAS可检测到与株高相关的基因[30]和[31], 并检测到基因, 该基因通过延迟开花进而影响株高, 而其他3种方法只能检测到基因。对于穗重, ALGWAS方法可检测到与产量相关的基因[32][33][34][35][36]和[37], 而其他3种方法几乎检测不到相关基因。通过QQ图, 可以发现ALGWAS相比于其他3种常用的混合线性模型方法均具有更高的统计功效, 同时对背景噪音导致的假阳性有较好的控制(图5)。ALGWAS方法检测到的已知基因位置及其对应的peakSNP位置见表2。
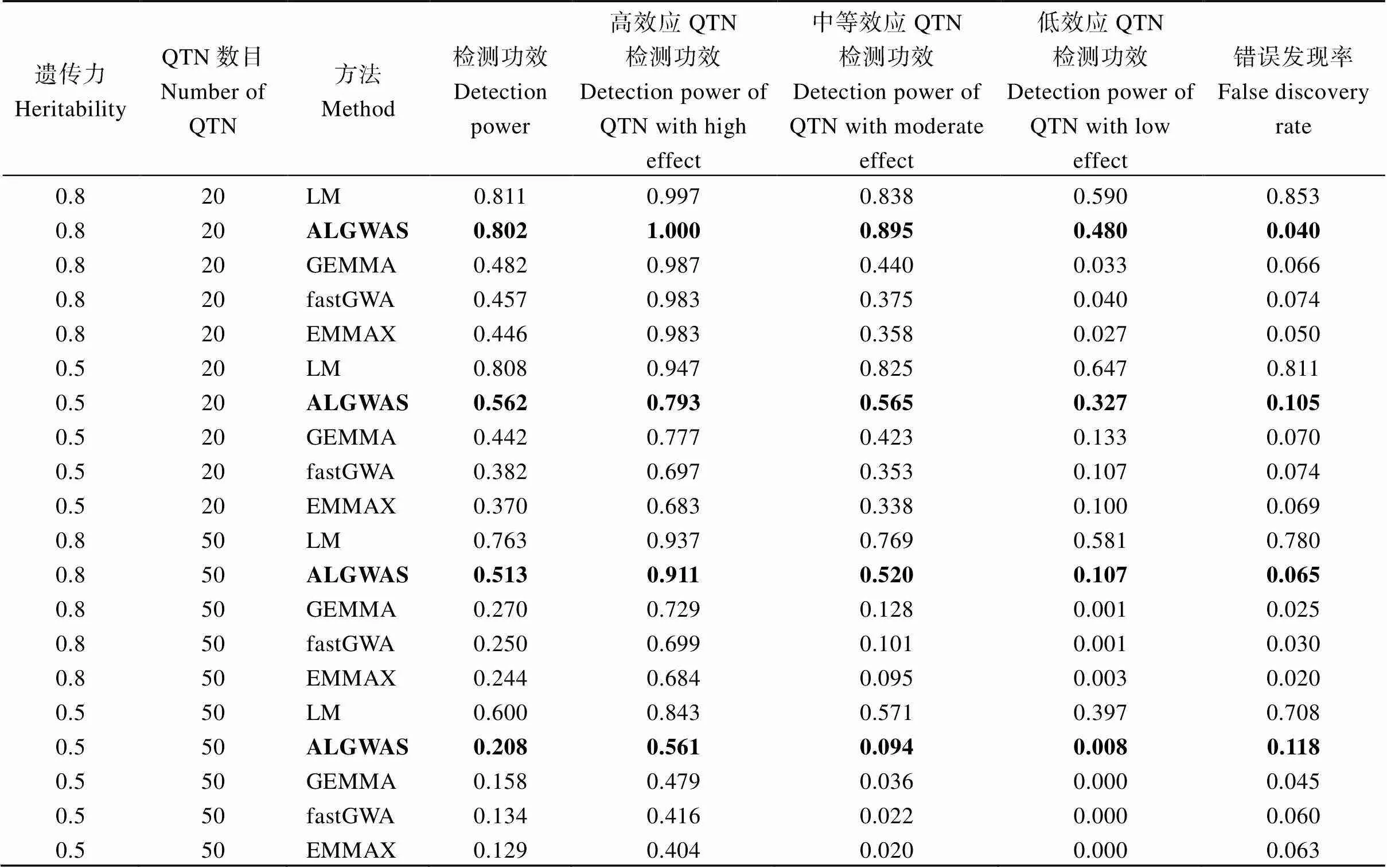
表1 基于从头模拟表型的不同全基因组关联分析方法的平均检测功效和错误发现率

图1 基于真实性状遗传结构模拟表型的不同全基因组关联分析方法的检测功效
A: 抽雄期; B: 株高; C: 穗重。A: days to tasseling; B: plant height; C: ear weight.

图2 CUBIC群体抽雄期的曼哈顿图
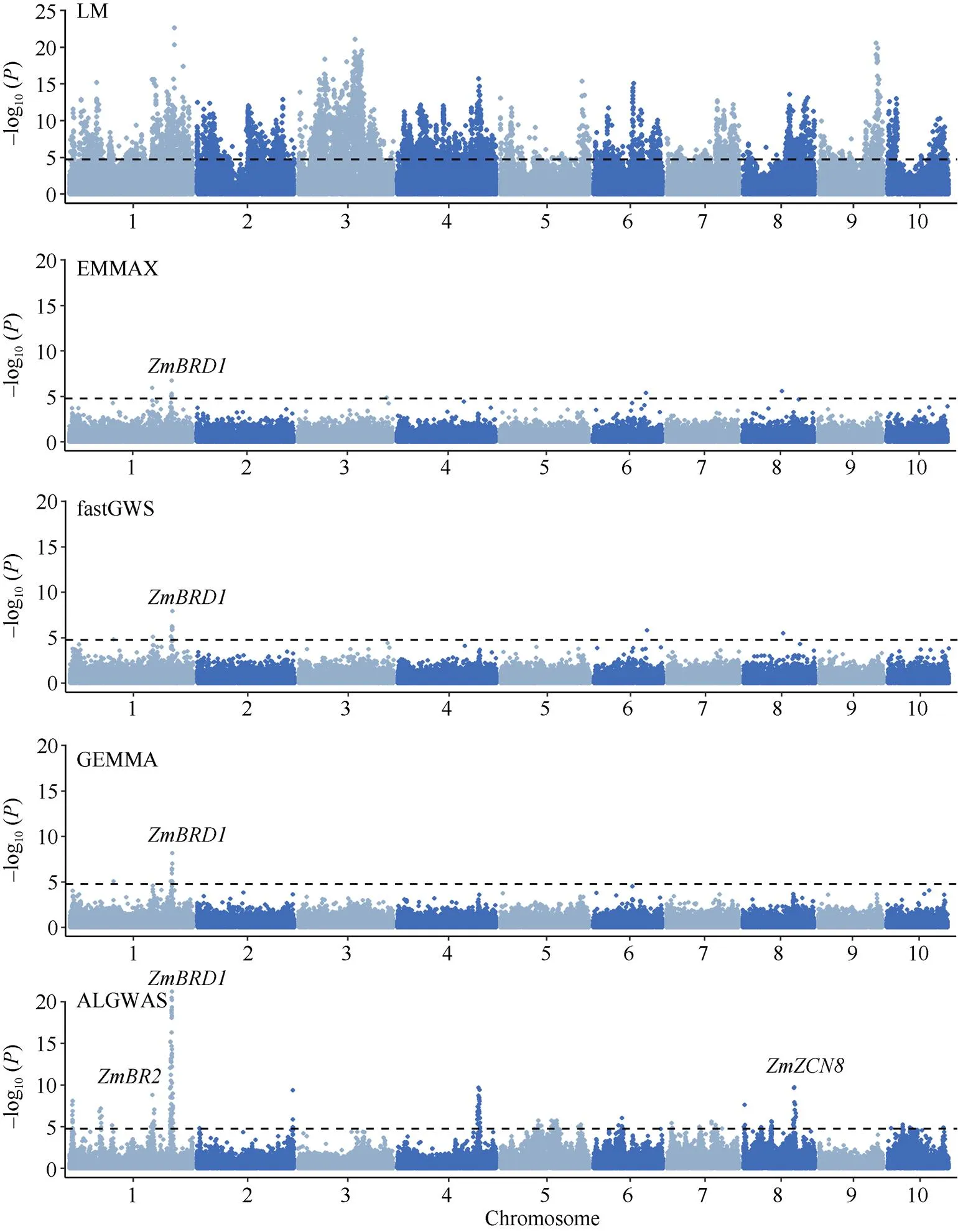
图3 CUBIC群体株高的曼哈顿图
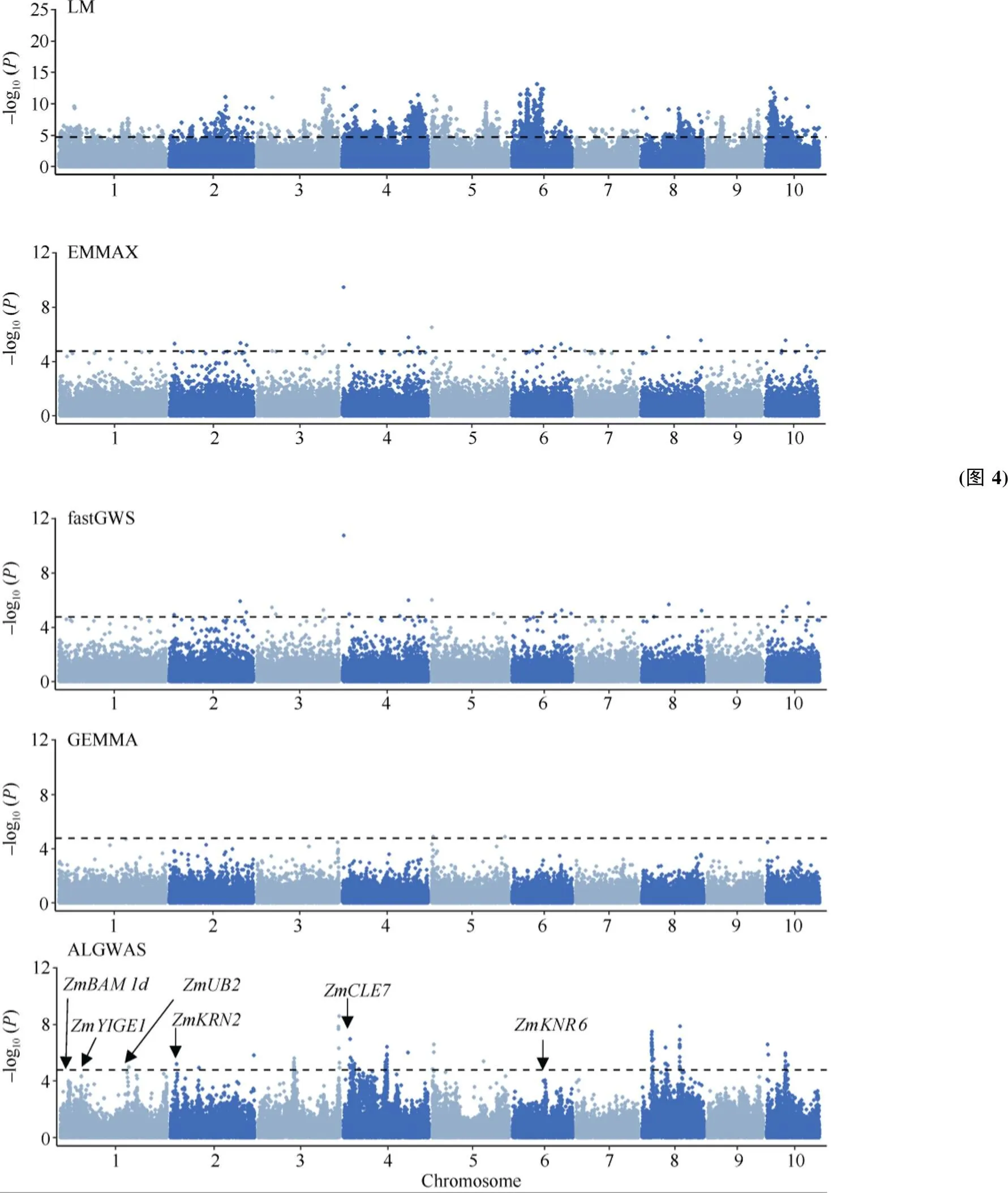
图4 CUBIC群体穗重的曼哈顿图

图5 不同全基因组关联分析方法的QQ图
A: 抽雄期; B: 株高; C: 穗重。A: days to tasseling; B: plant height; C: ear weight.
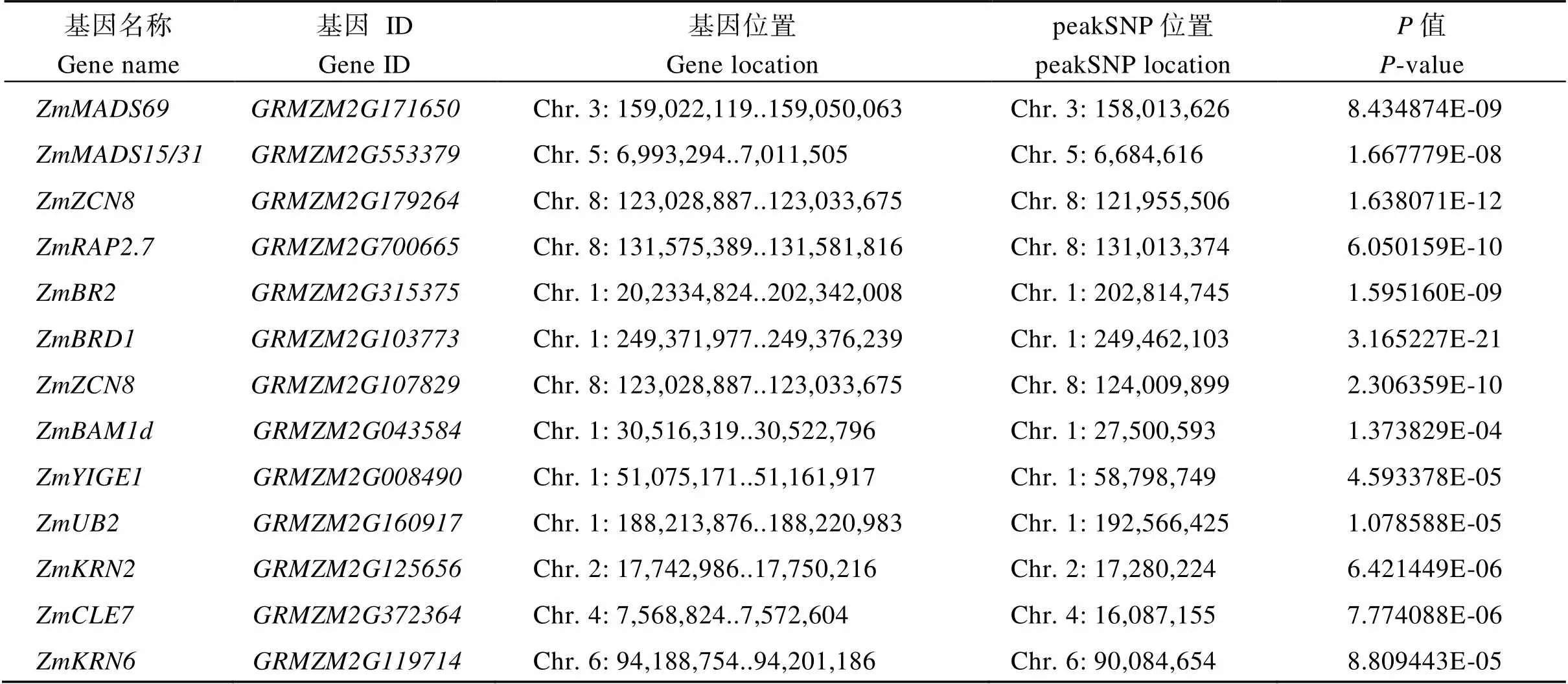
表2 ALGWAS方法检测到的已知基因位置及其对应的peakSNP位置
3 讨论
ALGWAS的第1阶段需要筛选与性状相关的SNP, 这一步可通过变量选择方法实现, 本研究选用的是Adaptive Lasso方法, 因为Zou给出了该方法具有一致性的理论证明[28]。本研究提供的是一个2阶段方法的框架, 其他的变量选择方法也可用于ALGWAS, 比如机器学习方法。在实际GWAS研究中, 如全基因组SNP数目达到百万级别时, ALGWAS的变量筛选阶段建议从中随机抽取一部分SNP作分析。
ALGWAS方法虽然在检测功效上具有优势, 但是它本身也有一定的局限性。ALGWAS的第2阶段进行单点扫描时, 每一次都需要对进入模型的协变量进行判断, 这一步导致了ALGWAS的速度还有待提高, 在后期的研究中, 我们将参考EMMAX[4]的做法, 通过固定进入模型的协变量来对其进行提速。
为了进一步提升ALGWAS方法的检测功效, 可以参考Li等[38]在CIM (Composite Interval Mapping)的基础上提出ICIM (Inclusive CIM)[39]的作法, 将ALGWAS第1阶段通过Adaptive Lasso方法得到的SNP优化权重直接用于第2阶段模型的学习。采用此方法也可进一步对ALGWAS方法进行提速。
4 结论
本研究提出了一种基于Adaptive Lasso的2阶段全基因组关联分析方法ALGWAS, 相比于目前常用的混合线性模型GWAS方法, ALGWAS在较好控制假阳性情况下, 统计功效更高, 特别对于产量等微效多基因遗传的性状, ALGWAS具有明显的检测优势, 这为复杂性状解析提供了新的解决途径。
[1] Zhang Y M, Mao Y C, Xie C Q, Smith H, Luo L, Xu S Z. Mapping quantitative trait loci using naturally occurring genetic variance among commercial inbred lines of maize (L.)., 2005, 169: 2267–2275.
[2] Yu J M, Pressoir G, Briggs H W, Vroh B I, Yamasakiet M, Doebley J F, McMullen M D, Gaut B S, Nielsen D M, Holland J B, Kresovich S, Buckler E S. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness., 2006, 38: 203–208.
[3] Kang H M, Zaitlen N A, Wade C M, Kirby A, Heckerman D, Daly M J, Eskin E. Efficient control of population structure in model organism association mapping., 2008, 178: 1709–1723.
[4] Kang H M, Sul J H, Service S K, Zaitlen N A, Kong S Y, Freimer N B, Sabatti C, Eskin E. Variance component model to account for sample structure in genome-wide association studies., 2010, 42: 348–354.
[5] Zhang Z W, Ersoz E, Lai C Q, Todhunter R J, Tiwari H K, Gore M A, Bradbury P J, Yu J, Arnett D K, Ordovas J M, Buckler E S. Mixed linear model approach adapted for genome-wide association studies., 2010, 42: 355–360.
[6] Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies., 2012, 44: 821–824.
[7] Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3000 shared controls., 2007, 447: 661–678.
[8] Li H, Peng Z Y, Yang X H, Wang W D, Fu J J, Wang J H, Han Y J, Chai Y C, Guo T T, Yang N, Liu J, Warburton M L, Cheng Y B, Hao X M, Zhang P, Zhao J Y, Liu Y J, Wang G Y, Li J S, Yan J B. Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels., 2013, 45: 43–50.
[9] Huang X H, Wei X H, Sang T, Zhao Q, Feng Q, Zhao Y, Li C Y, Zhu C R, Lu T T, Zhang Z W, Li M, Fan D L, Guo Y L, Wang A, Wang L, Deng L W, Li W J, Lu Y Q, Weng Q J, Liu K Y, Huang T,Zhou T Y, Jing Y F, Li W, Lin Z, Buckler E S, Qian Q, Zhang Q F, Li J Y, Han B. Genome-wide association studies of 14 agronomic traits in rice landraces., 2010, 42: 961–969.
[10] Xiao Y J, Liu H J, Wu L J, Warburton M L, Yan J B. Genome- wide association studies in maize: praise and stargaze., 2017, 10: 359–374.
[11] 彭勃, 赵晓雷, 王奕, 袁文娅, 李春辉, 李永祥, 张登峰, 石云素, 宋燕春, 王天宇, 黎裕. 玉米叶向值的全基因组关联分析. 作物学报, 2020, 46: 819–831. Peng B, Zhao X L, Wang Y, Yuan W Y, Li C H, Li Y X, Zhang D F, Shi Y S, Song Y C, Wang T Y, Li Y. Genome-wide association studies of leaf orientation value in maize., 2020, 46: 819–831 (in Chinese with English abstract).
[12] 谢磊, 任毅, 张新忠, 王继庆, 张志辉, 石书兵, 耿洪伟. 小麦穗发芽性状的全基因组关联分析. 作物学报, 2021, 47: 1891–1902. Xie L, Ren Y, Zhang X Z, Wang J Q, Zhang Z H, Shi S B, Geng H W. Genome-wide association study of pre-harvest sprouting traits in wheat., 2021, 47: 1891–1902 (in Chinese with English abstract).
[13] 杨飞, 张征锋, 南波, 肖本泽. 水稻产量相关性状的全基因组关联分析及候选基因筛选. 作物学报, 2022, 48: 1813–1821. Yang F, Zhang Z F, Nan B, Xiao B Z. Genome-wide association analysis and candidate gene selection of yield related traits in rice., 2022, 48: 1813–1821 (in Chinese with English abstract).
[14] Lippert C, Listgarten J, Liu Y, Kadiel C M, Davidson R I, Heckerman D. FaST linear mixed models for genome-wide association studies., 2011, 8: 833–835.
[15] Listgarten J, Lippert C, Kadie C M, Davidson R I, Eskin E, Heckerman D. Improved linear mixed models for genome-wide association studies., 2012, 9: 525–526.
[16] Loh P R, Bhatia G, Gusev A, Finucane H K, Bulik-Sullivan B K, Pollack S J. Contrasting genetic architectures of schizophrenia and other complex diseases using fast variance-components analysis., 2015, 47: 1385–1392.
[17] Jiang L D, Zheng Z L, Qi T, Kemper K E, Wray N R, Visscher P M, Yang J. A resource-efficient tool for mixed model association analysis of large-scale data., 2019, 51: 1749–1755.
[18] Maher B. Personal genomes: the case of the missing heritability., 2008, 456: 18–21.
[19] Visscher P. Sizing up human height variation., 2008, 40: 489–490.
[20] Yang J, Benyamin B, McEvoy B P, Gordon S, Henders A K, Nyholt D R, Madden P A, Heath A C, Martin N G, Montgomery G W, Goddard M E, Visscher P M. Common SNPs explain a large proportion of the heritability for human height., 2010, 42: 565–569.
[21] Song B, Mott R, Gan X. Recovery of novel association loci inandthrough leveraging INDELs association and integrated burden test., 2018, 14: e1007699.
[22] Zhang Y W, Tamba C L, Wen Y J, Li P, Ren W L, Ni Y L, Gao J, Zhang Y M. mrMLM v4.0.2: an R platform for multi-locus genome-wide association studies., 2020, 18: 481–487.
[23] Yang N, Lu Y L, Yang X H, Huang J, Zhou Y, Ali F H, Wen W W, Liu J, Li J S, Yan J B. Genome wide association studies using a new nonparametric model reveal the genetic architecture of 17 agronomic traits in an enlarged maize association panel., 2014, 10: e1004573.
[24] Liu H J, Wang X Q, Xiao Y J, Luo J Y, Qiao F, Yang W Y, Zhang R Y, Meng Y J, Sun J M, Yan S J, Peng Y, Niu L Y, Jian L M, Song W, Yan J L, Li C H, Zhao Y X, Liu Y, Warburton M L, Zhao J R, Yan J B. CUBIC: an atlas of genetic architecture promises directed maize improvement., 2020, 21: 20.
[25] Lande R, Thompson R. Efficiency of marker-assisted selection in the improvement of quantitative traits., 1990, 124: 743–756.
[26] Yu J M, Holland J B, McMullen M D, Buckler E S. Genetic design and statistical power of nested association mapping in maize., 2008, 178: 539–551.
[27] Tibshirani R. Regression shrinkage and selectionthe lasso., 1996, 58: 267–288.
[28] Zou H. The adaptive lasso and its oracle properties., 2006, 101: 1418–1429.
[29] Liang Y M, Liu Q, Wang X F, Huang C, Xu G H, Hey S, Lin H Y, Li C, Xu D Y, Wu L S, Wang C L, Wu W H, Xia J L, Han X, Lu S J, Lai J S, Song W B, Schnable P S, Tian F. ZmMADS69 functions as a flowering activator through the regulatory module and contributes to maize flowering time adaptation., 2019, 221: 2335–2347.
[30] Makarevitch I, Thompson A, Muehlbauer G J, Springer N M.gene in maize encodes a brassinosteroid C-6 oxidase., 2012, 7: e30798.
[31] Xing A Q, Gao Y F, Ye L F, Zhang W P, Cai L C, Ching A, Llaca V, Johnson B, Liu L, Yang X H, Kang D M, Yan J B, Li J S. A rare SNP mutation in Brachytic2 moderately reduces plant height and increases yield potential in maize., 2015, 66: 3791–3802.
[32] Yang N, Liu J, Gao Q, Gui S T, Chen L, Yang L F, Huang J, Deng T Q, Luo J Y, He L J, Wang Y B, Xu P W, Peng Y, Shi Z, Lan L, Ma Z Y, Yang X, Zhang Q Q, Bai M Z, Li W, Liu L, Jackson D, Yan J B. Genome assembly of a tropical maize inbred line provides insights into structural variation and crop improvement., 2019, 51: 1052–1059.
[33] Luo Y, Zhang M L, Liu Y, Liu J, Li W Q, Chen G S, Peng Y, Jin M, Wei W J, Jian L M, Yan J, Fernie A R, Yan J B. Genetic variation in YIGE1 contributes to ear length and grain yield in maize., 2022, 234: 513–526.
[34] Du Y F, Liu L, Peng Y, Li M F, Li Y F, Liu D, Li X W, Zhang Z X.expression and inflorescence development is mediated byand the distal enhancer,, in maize., 2020, 16: e1008764.
[35] Chen W K, Chen L, Zhang X, Yang N, Guo J H, Wang M, Ji S G, Zhao X Y, Yin P F, Cai L C, Xu J, Zhang L L, Han Y J, Xiao Y N, Xu G, Wang Y B, Wang S H, Wu S, Yang F, Jackson D, Cheng J K, Chen S H, Sun C Q, Qin F, Tian F, Fernie A R, Li J S, Yan J B, Yang X H. Convergent selection of a WD40 protein that enhances grain yield in maize and rice., 2022, 375: e7985.
[36] Liu L, Gallagher J, Arevalo E D, Chen R, Skopelitis T, Wu Q, Bartlett M, Jackson D. Enhancing grain-yield-related traits by CRISPR-Cas9 promoter editing of maize CLE genes., 2021, 7: 287–294.
[37] Jia H T, Li M F, Li W Y, Liu L, Jian Y N, Yang Z X, Shen X M, Ning Q, Du Y F, Zhao R, Jackson D, Yang X H, Zhang Z X. A serine/threonine protein kinase encoding gene KERNEL NUMBER PER ROW6 regulates maize grain yield., 2020, 11: 988.
[38] Zeng Z B. Precision mapping of quantitative trait loci.1994, 136: 1457–1468.
[39] Li H H, Ye G Y, Wang J K. A modified algorithm for the improvement of composite interval mapping., 2007, 175: 361–374.
ALGWAS: two-stage Adaptive Lasso-based genome-wide association study
YANG Wen-Yu1,2, WU Cheng-Xiu1, XIAO Ying-Jie1,3,*, and YAN Jian-Bing1,3
1National Key Laboratory of Crop Genetic Improvement, Huazhong Agricultural University, Wuhan 430070, Hubei, China;2College of Science, Huazhong Agricultural University, Wuhan 430070, Hubei, China;3Hubei Hongshan Laboratory, Wuhan 430070, Hubei, China
As mainstream methods for genome-wide association analysis, mixed linear model methods have been widely used. However, the existing methods still have the problem of low detection power. In this study, a two-stage Adaptive Lasso-based genome-wide association analysis (ALGWAS) method was proposed. In the first stage, single nucleotide polymorphism (SNP) associated with target traits were screened by Adaptive Lasso, a variable selection method. In the second stage, SNPs selected from the first stage were put into the linear model as the covariates for genome-wide scanning. Compared with fastGWA, GEMMA and EMMAX, the ALGWAS method had the highest detection power and lower false discovery rate (FDR) in the simulation experiments. The above four methods were applied to genome-wide association analysis of Complete-diallel plus Unbalanced Breeding-like Inter-Cross (CUBIC) population of 1341 individuals in maize. ALGWAS method can detect the genes (,,, andrelated to days to tasseling, the genes (and) related to plant height, and the genes (,,and) related to yield, while the other three commonly used genome-wide association analysis methods had low detection efficiency. In this study, a non-mixed linear model class of genome-wide association analysis method was proposed, which had higher detection advantage for microeffect polygenes and provided a new way for genetic analysis of complex traits.
maize; genome-wide association study; variable selection; Adaptive Lasso
2022-10-28;
2023-02-21;
2023-03-03.
10.3724/SP.J.1006.2023.23072
通信作者(Corresponding author):肖英杰, E-mail: yxiao25@mail.hzau.edu.cn
E-mail: yangwenyurain@126.com
本研究由国家自然科学基金项目(32201855, 32122066)资助。
This study was supported by the National Natural Science Foundation of China (32201855, 32122066).
URL: https://kns.cnki.net/kcms/detail/11.1809.S.20230302.1544.007.html
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
