甲哌鎓原药及其制剂的高效液相亲水色谱分析
2023-07-19王东连严艳伦姜治国
王东连,张 杰,严艳伦,姜治国
(云南云大科技农化有限公司,云南 昆明 650000)
甲哌鎓,ISO通用名称:mepiquat chloride。其他中文名称:助壮素、缩节胺、调节啶、甲哌啶、壮棉素、缩节灵。化学名称:N,N-二甲基哌啶鎓氯化物。分子式:C7H16NCl。相对分子质量:149.66 (按2007年相对原子质量计)。具有植物生长调节作用。22.5% 28-表芸·甲哌鎓水剂是由甲哌鎓、28-表高芸苔素内酯复配而成的棉花株铃调控剂。药效实验结果表明,甲哌鎓和28-表高芸苔素内酯复配后可显著促进根系生长,健株稳长、塑造株型、改善叶片功能和群体结构,最终达到优化成铃结构、增加铃数和铃重的功效,对棉花增产有显著的增效作用。本品用于调控棉花株型、防止蕾铃脱落、提高产量,对于解决棉花生产中徒长和蕾铃脱落的问题有广阔的应用前景。迄今为止,大多数文献和相关标准所用的检验方法大致为离子对色谱柱与电导检测器结合、离子对试剂与反相色谱柱和紫外检测器结合、电位滴定与纸层析结合、液质联用等。现有检测方法,大部分不能在色谱柱上很好的保留。电位滴定法又测定步骤多;花费时间长;灵敏度低,对制剂产品的准确性更差。经查阅甲哌鎓化学结构和相关性质后,决定采用近几年快速发展的亲水色谱技术与VWD或DAD检测器结合的方式进行分离检测。该色谱技术即使是强极性的化合物也能通过HILIC使用与反相HPLC相同的系统和溶剂实现分离,大大降低了极性分析的成本和工作量。
1 试验部分
1.1 试剂和溶液 甲哌鎓标准品:质量分数99.2%;22.5% 28-表芸·甲哌鎓水剂:是由甲哌鎓、28-表高芸苔素内酯复配而成。乙腈:色谱纯,飞世尔;乙酸铵:色谱纯;试验用水:去离子水。
1.2 仪器 高效液相色谱仪(带DAD检测器):安捷伦1260 II;色谱柱:默克ZIC©-HILIC,2.1×100 mm,3.5 μm;电子分析天平:梅特勒AB204-E;超声波清洗机:新芝SB25-12DTD。
1.3 高效液相色谱操作条件 流动相:80%的乙腈+20%的25 mmol/L乙酸铵水溶液;流速:0.2 mL/min;柱温:30℃;检测波长:210 nm;进样体积:2 μL;保留时间:约为4.18 min左右。
1.4 测定步骤
1.4.1 标准溶液的配制 准确称取甲哌鎓标准样品0.10 g(精确到0.000 2 g),用80%的乙腈+20%的25 mmol/L乙酸铵水溶液溶解,并定容至25 mL容量瓶中。摇匀,超声波脱气15 min,过0.22 μm的尼龙膜至1.5 mL进样瓶中,备用。
1.4.2 原药及制剂样品溶液的配制
1.4.2.1 原药测定溶液的配制 准确称取甲哌鎓原药0.10 g(精确到0.000 2 g),用80%的乙腈+20%的25 mmol/L乙酸铵水溶液溶解,并稀释至25 mL容量瓶中,摇匀,超声波脱气15 min,过0.22 μm的尼龙膜至1.5 mL进样瓶中,备用。
1.4.2.2 水剂测定溶液的配制 用已测定甲哌鎓原药按照工艺配方制成一个待测样品,同时制备一个制剂空白。然后,准确称取含0.10 g甲哌鎓的水剂样品(精确至0.000 2 g)于25 mL容量瓶中,用流动相溶解并稀释至刻度,摇匀,超声波脱气,过滤备用。
1.4.3 测定 在上述色谱条件下,待仪器稳定后,先注入数针标样溶液,计算各针峰面积,直至相邻2针标样溶液峰面积变化<1.5%后,按照标样、试样、试样、标样的顺序进样测定。
1.4.4 计算 将测得的2两针试样溶液以及试样前后2针溶液中甲哌鎓的峰面积,分别进行平均。甲哌鎓的质量分数X1(%)按公式(1)进行计算:
(1)
式中:
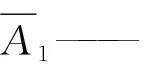
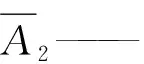
m1——标准品甲哌鎓的质量,g;
m2——试样的质量,g;
P1——标准品甲哌鎓的质量分数,%。
2 结果与分析
2.1 色谱条件的选择 甲哌鎓属于强极性化合物,在液相分离中使用反相柱或者阳离子柱,保留时间较短且易受到溶剂峰的干扰,很难达到分析定量的目的。其吸收最佳波长195 nm[1],但195~220 nm处吸收较强。用安捷伦亲水色谱柱发现波长增大灵敏度降低,柱压变化较大,柱子耐用性明显下降。且未得到高灵敏度,检测波长选择较低。后经过调研,发现默克ZIC©-HILIC柱带有PEEK内衬[2],能很好的降低极性化合物在柱子上的残留。经过多次试验,甲哌鎓在默克ZIC©-HILIC柱上不仅灵敏度高,而且在210 nm波长处也有很好的响应及线性,同时也能排除低波长的溶剂效应。所以将波长选择为210 nm。亲水色谱柱[2],即使是强极性的化合物也能通过HILIC使用与反相HPLC相同的系统和溶剂实现分离,大大降低了极性分析的成本和工作量,且提高了分离度。此液相条件下分离,目标物不受杂质峰干扰。首先,用乙腈-水95+5→60+40做梯度看甲哌鎓在柱子上的保留情况。结果显示保留较好。再次,用乙腈-水90+10和80+20两个等度对目标物进行洗脱看保留时间。结果表明,当80+20等度洗脱时能节约时间且与干扰峰很好地分离。第三,由于峰的对称性较差,所以选择加入缓冲盐来改善峰型。首选甲酸铵与乙酸铵。本实验直接采用乙酸铵,在25 mmol/L浓度时能很好的得到保留和分离(图1)。
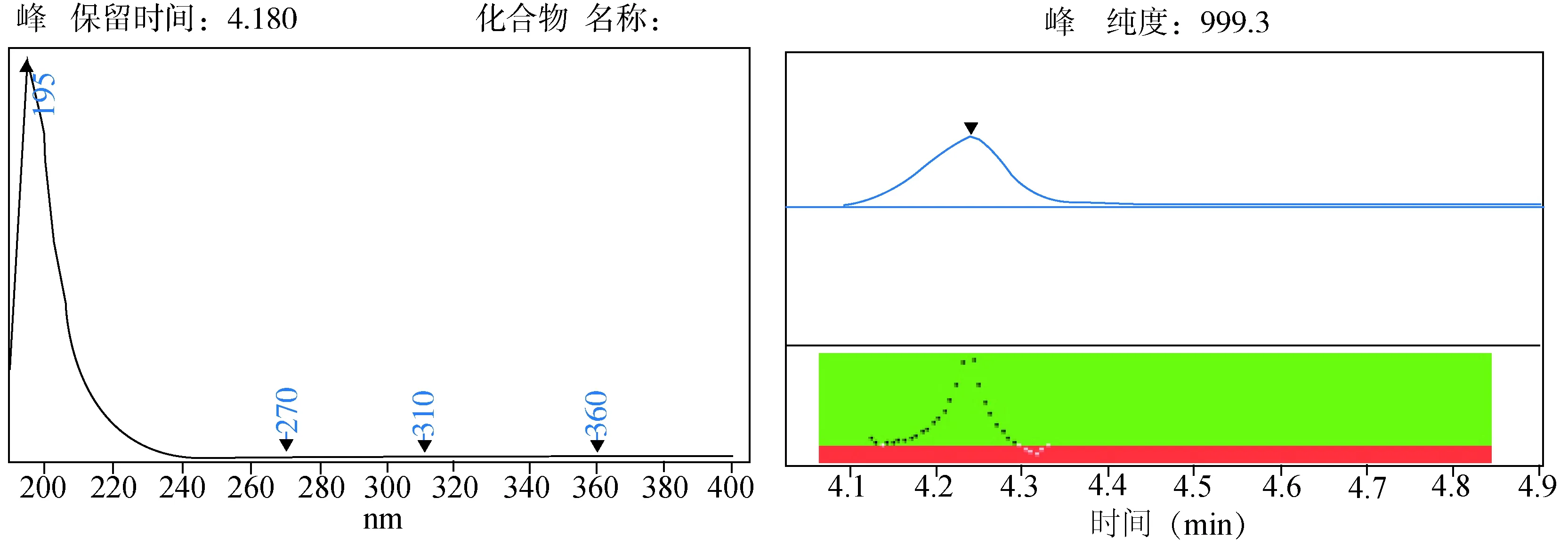
图1 甲哌鎓紫外光谱图
2.2 分析方法线性相关性试验 标准溶液分别进样1~5 μL。在上述色谱条件下进行测定,以进样量为纵坐标y,以峰面积为横坐标x,绘制标准曲线,得到线性方程为y=0.002x-0.144 6,相关系数r=0.998 9[3]。甲哌鎓的线性相关性实验结果(表1),线性关系(图2)。
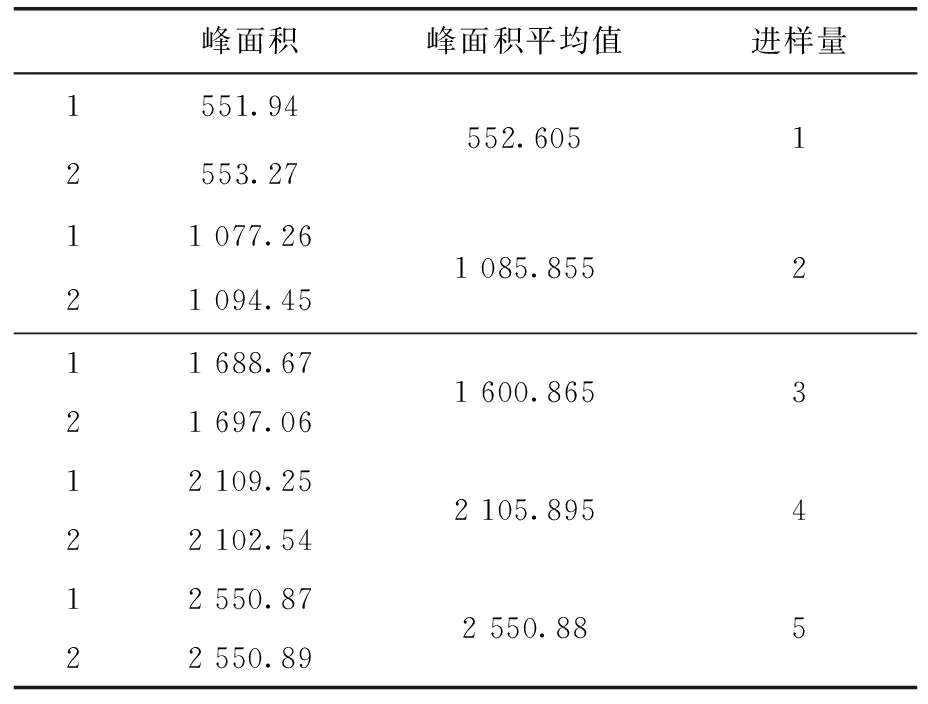
表1 线性相关实验结果
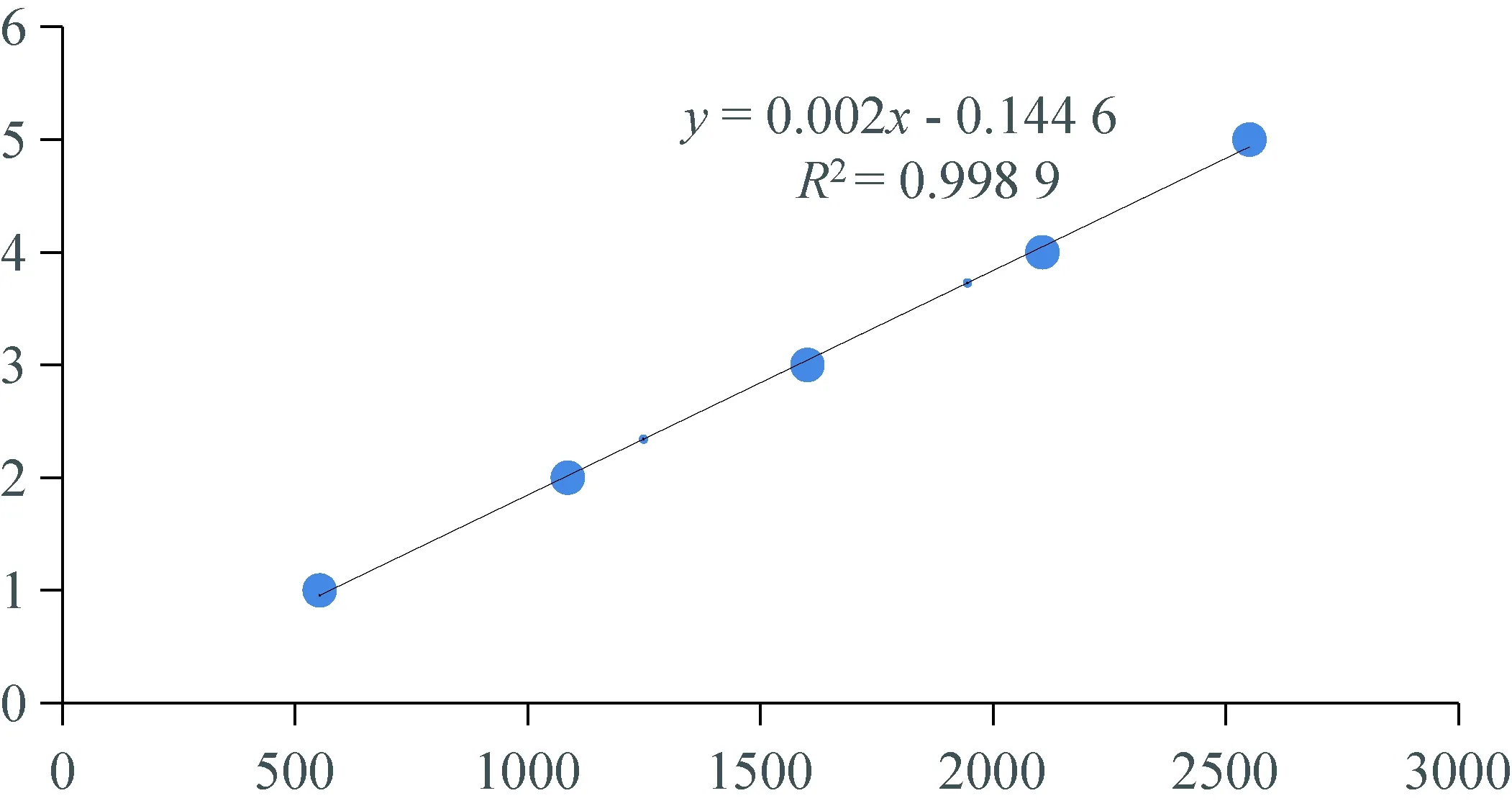
图2 甲哌鎓线性关系图
2.3 分析方法的精密度试验 在相同的色谱条件下,从同一产品中准确称取5个试样,在上述色谱条件下进行分析并求平均值、标准偏差和变异系数,结果表明:甲哌鎓的标准偏差为:0.209 6,变异系数为:0.89%(表2)。

表2 分析方法的精密度试验结果
2.4 分析方法的准确度试验 试验采用产品空白制剂中添加已知质量分数的试样5份,按照1.4进行分析,结果表明:甲哌鎓的平均回收率为:99.8%(表3)。
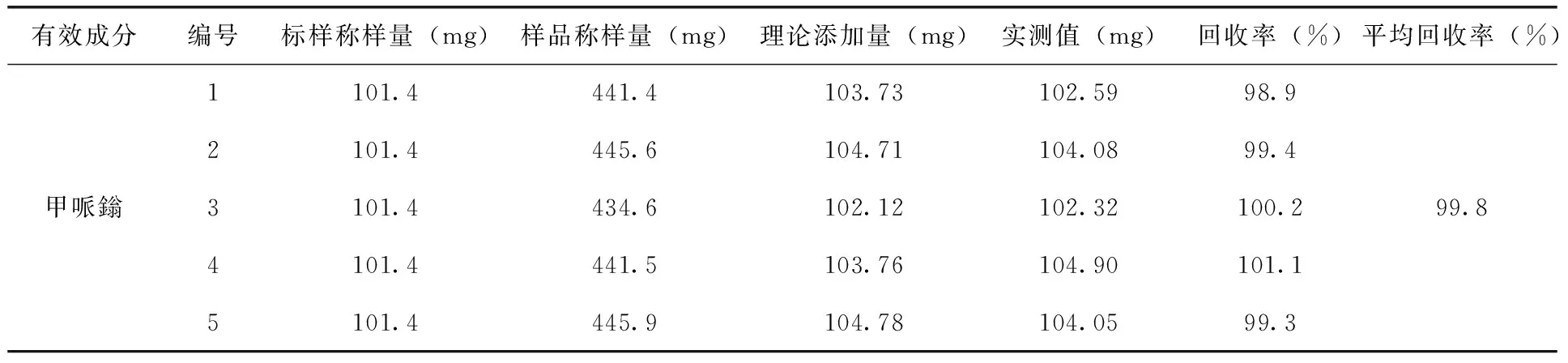
表3 分析方法的准确度试验结果
2.5 非分析物干扰的判定 在评价准确度时,需要包含非分析物质干扰试验,因为助剂中的任何干扰物质都会导致分析方法出现偏差,分析时同时测定不带助剂的原药和空白样品,证明其无干扰,并提交色谱图。通过不加助剂的原药色谱图和空白样品色谱图判定,非分析物对有效成分不构成干扰,相关非分析物质干扰典型图谱(图3~7)。
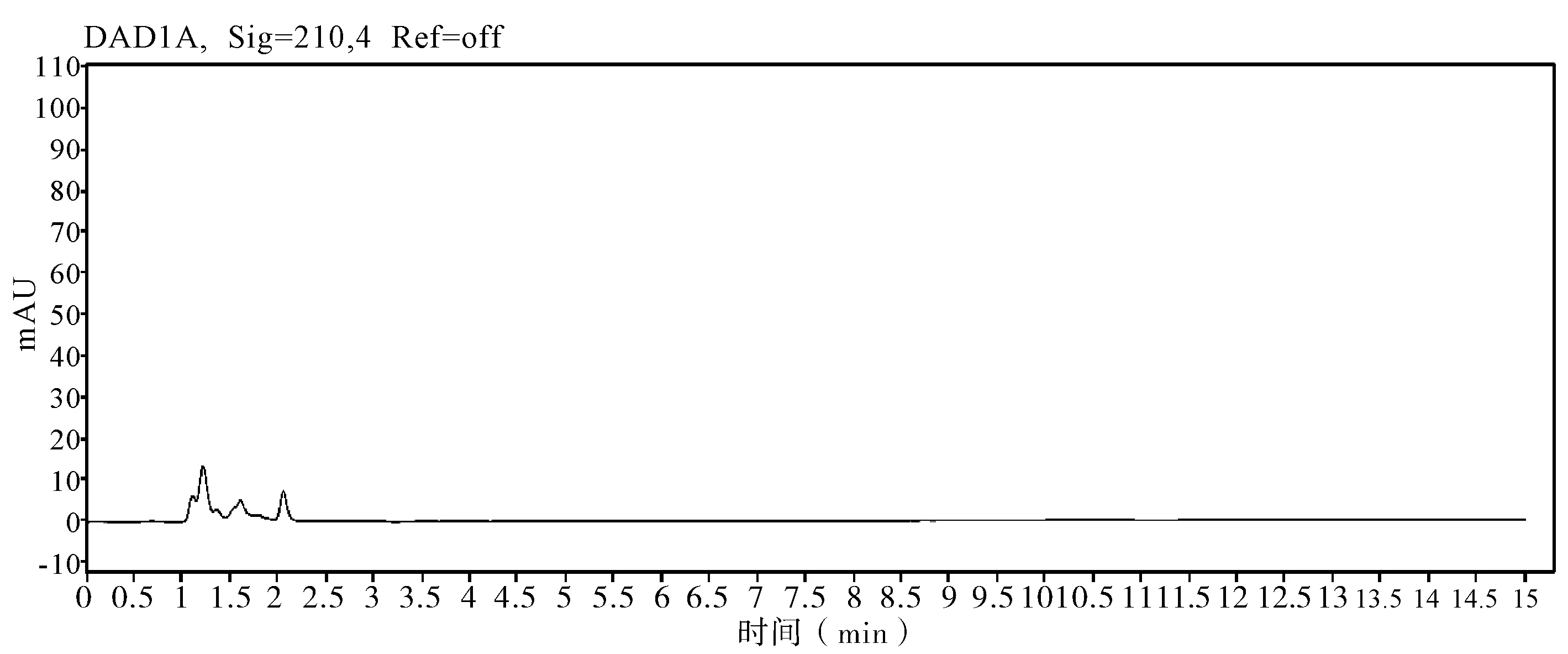
图3 溶剂空白
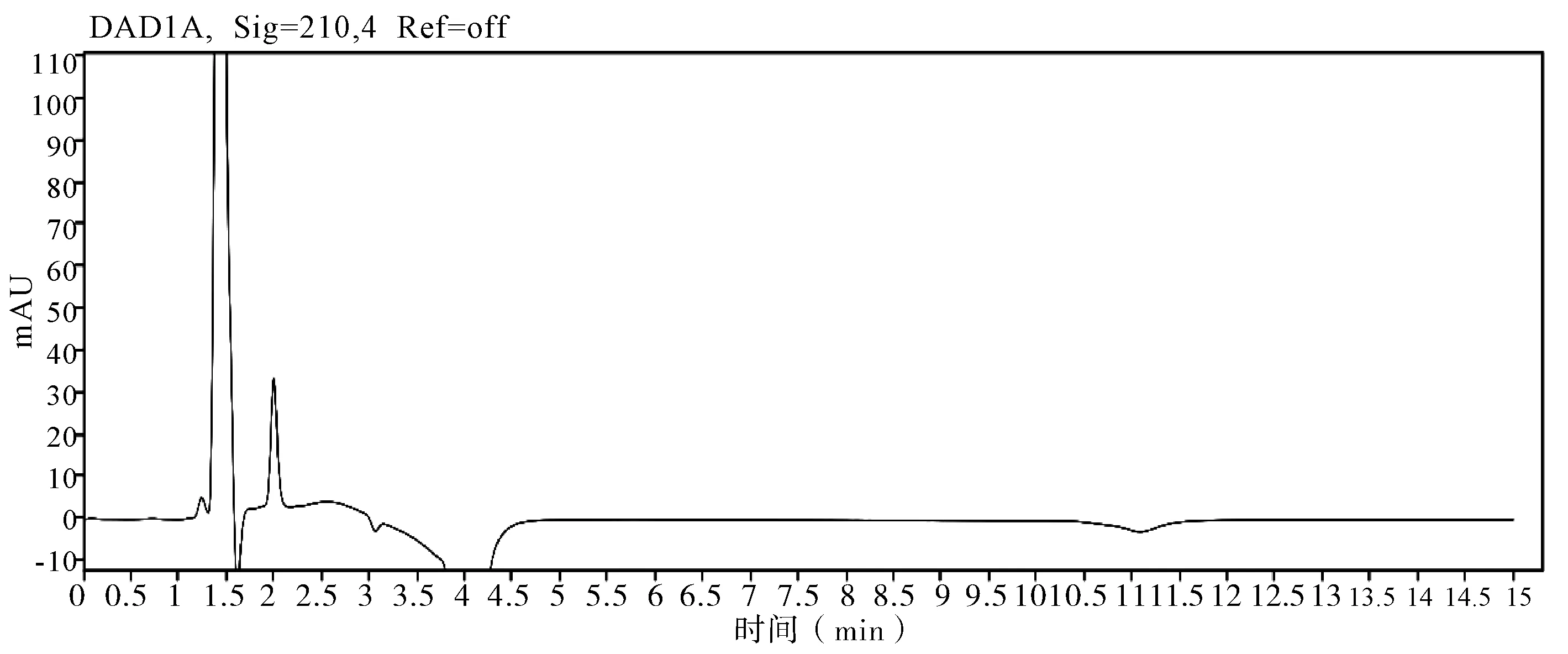
图4 制剂空白
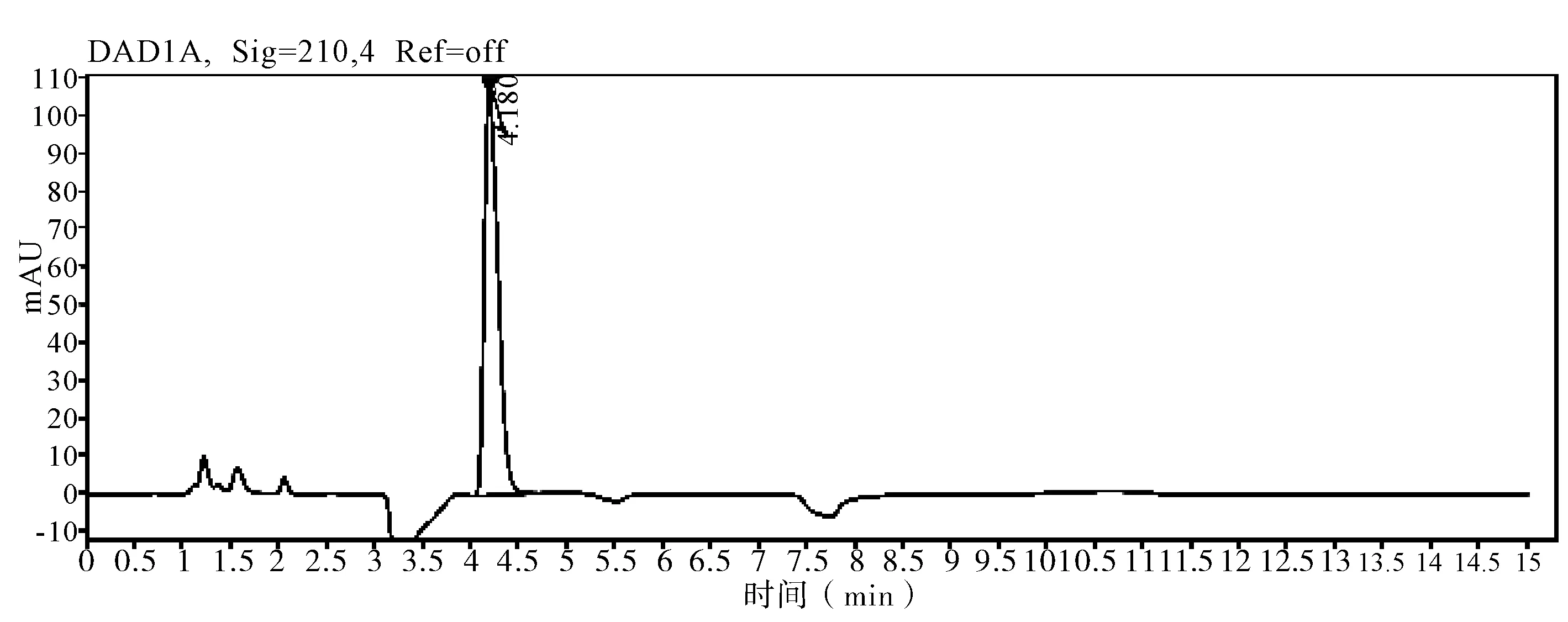
图5 标样图谱
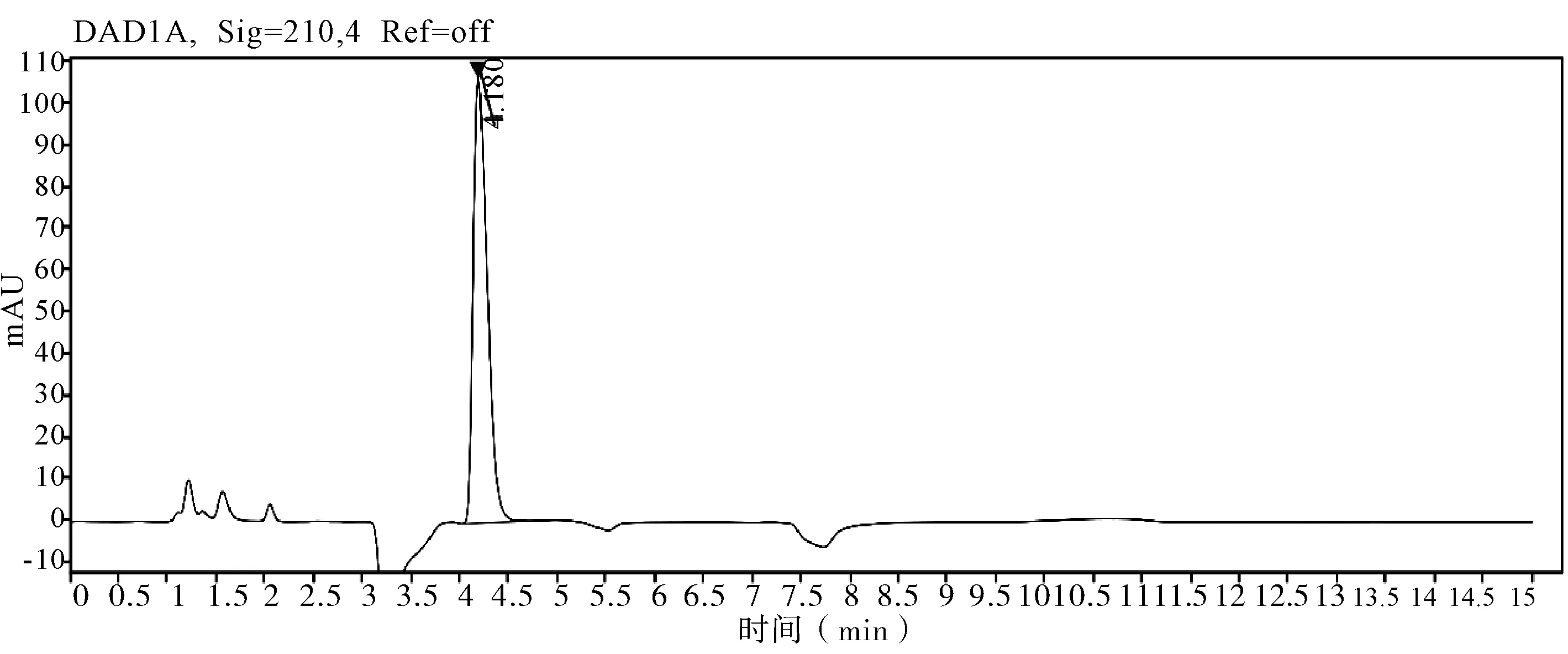
图6 甲哌鎓水剂图谱
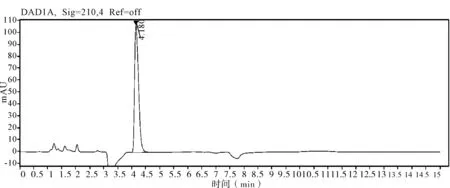
图7 甲哌鎓原药图谱
3 结论
综上所述,用本分析方法对甲哌鎓原药及22.5% 28-表芸·甲哌鎓水剂进行测定分析,方法准确度高,精密度高,分析方法简便、快速,适用于甲哌鎓原药及22.5% 28-表芸·甲哌鎓水剂产品的质量控制分析。
