长链非编码RNA共表达基因在卵巢上皮性癌的生物学作用
2023-05-30李耀威寿坚陈龙
李耀威 寿坚 陈龙
卵巢癌(ovarian cancer,OC)是导致妇科恶性肿瘤患者死亡的主要原因,75%OC病理类型系上皮性来源,卵巢上皮性癌(epithelial ovarian carcinoma,EOC)患者的生存率近年来未明显提高[1],鉴定敏感且切实有效的生物标志物对实现早期诊断或有效预测EOC 患者的临床预后有重要的现实意义。lncRNA是一类长度超过200个核苷酸的非编码RNA,越来越多证据表明lncRNA 起着癌基因、抑癌基因或两者兼备的作用[2-4]。然而,大多数lncRNA 的表达模式、生物学功能和临床意义仍不甚清楚。本研究对在OC 患者中表达异常lncRNA 相关联的蛋白质编码基因(protein coding gene,PCG)进行生物信息学分析,以期对lncRNA 在参与OC 致病、进展、预后等方面机制增进了解,进而为后续实验设计提供线索。
1 资料与方法
1.1 获取OC 组织中与lncRNA 共表达且差异表达的基因(1)差异表达mRNA 芯片数据的获取:①在美国国立生物技术中心(NCBI)的Gene Expression Omnibus(GEO)数据库中检索与OC 相关的mRNA 表达谱芯片数据集(检索条件:研究类型为expression profiling by array、种属为homo sapiens、病例和对照样本数目均≥10 例、时间为自建库至2022 年12 月31 日),随后下载符合纳入条件的mRNA 表达谱芯片数据集备后续分析。②R/Bioconductor 中的Limma 软件包(3.36.5 版)用于识别OC组织和正常卵巢上皮组织之间的差异表达基因(DEG)。使用Benjamini 和Hochberg 提出的伪发现率(FDR)得到调整后P值纠正伪阳性结果。P<0.05和|log2(FC)|>1 设置为差异基因的纳入标准[注:FC表示差异倍数(fold change)]。根据下载的平台注释文件匹配矩阵文件中的原始探针数据为基因名称,通过最小P 值选择同一基因对应的多个探针的表达值作为该基因的表达值。(2)在OC 组织中与lncRNA 共表达基因的获取。①利用关键词“long non-coding RNA”、“long noncoding RNA”、“lncRNA”、“ovarian cancer”、“ovarian carcinoma”、“ovarian neoplasm”、“ovarian tumor”、“ovarian tumors”、“ovarian tumour”、“ovarian tumours”、“ovarian malignancy”通过计算机及手工检索Medline/PubMed、EMBASE、Web of Knowledge 数据库,检索时间从建库至2022 年12 月31 日。查找来源于OC 患者、经过实验证实表达异常且明确已知其序列及结构等注释信息的lncRNA 纳入分析。②利用perl 语言及R 语言平台使用皮尔森相关系数和z-test 检验目标lncRNA 的表达水平与每个PCG 之间的相关性。与目标lncRNA 正或负相关的PCG 被视为与lncRNA 相关的PCG(| pearson correlation|> 0.4,P<0.01 为判定标准)。(3)通过Venny 2.1.0 在线工具,取相关芯片数据差异基因(A)与lncRNA 共表达相关基因(B)的交集即获得OC 组织中与lncRNA 共表达且差异表达的PCG。
1.2 生物学功能及通路富集分析 利用在线数据库DAVID 中GO 和KEGG 进行生物功能及通路富集分析,FDR<0.05 判定为有统计学意义。
1.3 PPI 网络构建 及hub gene 确 定 PPI 网络由STRING 数据库构建,并使用Cytoscape 进行可视化处理。Hub gene 是在生物学过程中发挥至关重要作用的基因,在相关通路中,其他基因的调控常受该基因的影响,PPI 网络中degree ≥10 判定为hub gene 的纳入标准。
1.4 Module 分析 使用Cytoscape 软件MCODE 软件包进行module 分析,设定degree cutoff=2,node score cutoff=0.2,k-core=2,and max.depth=100。使用DAVID对module 中的DEG 进行GO 分析及KEGG 通路富集分析。
1.5 对hub gene 进行生存分析 OncoLnc 是与mRNA、miRNA 或lncRNA 的表达数据相关联的可用于生存分析的在线工具。
2 结果
2.1 OC 组织中与lncRNA 共表达且差异表达基因的获得(1)OC 组织中差异表达基因的获得:由GEO 数据库获得GSE14407 和GSE18520 两个mRNA 表达谱芯片数据集。GSE14407 和GSE1852 芯片数据集分别由12、53 个上皮性OC 组织和12、10 个正常卵巢上皮组织构成。从GSE14407、GSE18520 数据集中分别识别出2328 和9590 个DEG。(2)lncRNA 数据的获得:通过检索文献共获得9 种lncRNA(分别是LINC01088[5]、SNHG3[6]、SPRY4-IT1[7]、CPS1-IT1[8]、CDKN2BAS1(又 名ANRIL)[9]、MALAT1[10]、FAM215A[11]、LINC00472[11]和HOTAIR[12],以上均已知序列及结构等注释信息)供作者进行生物信息学分析研究。利用皮尔森相关系数和z-test 检验9 种lncRNA 的表达水平与每个PCG 之间的相关性后发现,9 种lncRNA 共表达的PCG 数目(去重后)总和为15,965 个。(3)利用在线工具venny 将GSE18520、GSE14407 数据集所得DEG同与lncRNA 共表达的PCG 取交集得到与lncRNA 共表达且属差异表达的基因共1,421 个。
2.2 与lncRNA 共表达且属差异表达基因的生物学功能分析 GO分析发现许多共表达差异基因参与了DNA replication、cell division、cell proliferation、extracellular exosome 及protein binding 等功能富集过程;KEGG 分析发现在这些共表达基因中有49 个基因参与了pathways in cancer 信号通路。见表1。
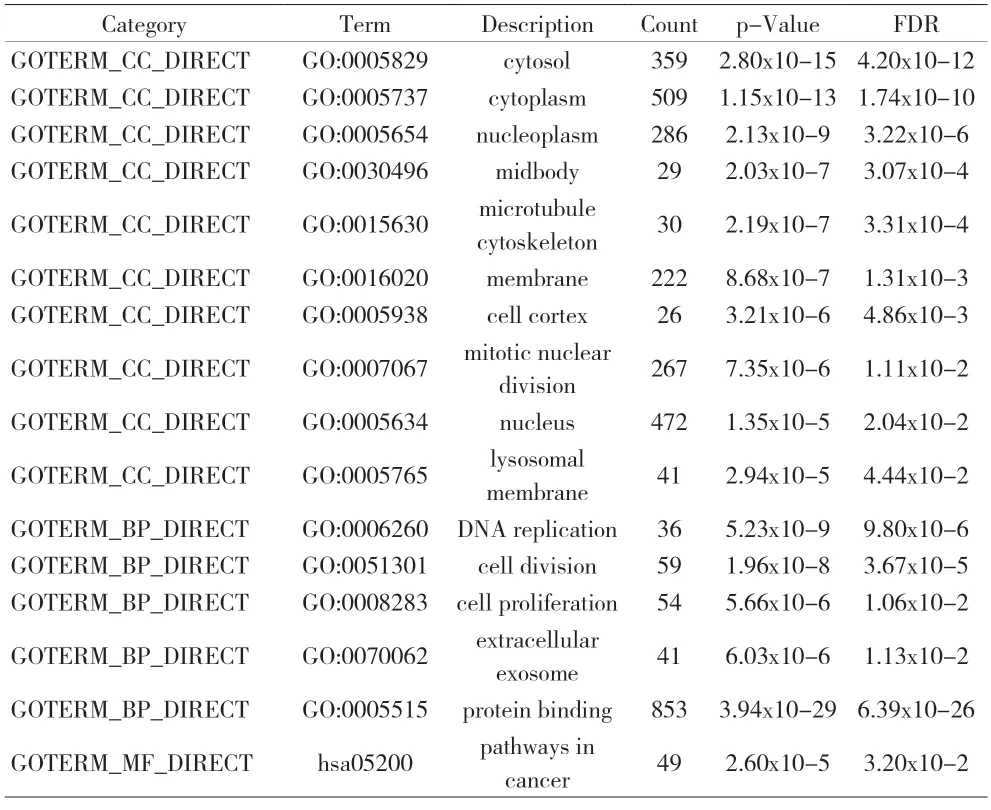
表1 与lncRNA共表达的差异表达基因的GO及KEGG分析
2.3 PPI 网络构建和hub gene 确定及互作分析 经PPI网络构建后,满足与lncRNA 共表达且属差异表达基因的PPI 网络由979 nodes 和5,060 edges 组成。随后筛选出满足条件的hub gene 共274 个。
2.4 Module 确定和功能富集分析 用Cytoscape 软件中的MCODE 应用程序分析互作网络后,获得2 个重要module,标记为module 1 和module 2,分别包括46、35个nodes 和917、290 个edges。对module 1 进行GO 分析表明,这些基因参与cell cycle、cell division、ATP binding、nucleoside binding、nucleotide binding、microtubule motor activity 等生物学过程;KEGG 分析发现参与Cell cycle及Oocyte meiosis 信号通路。对 module 2 进行GO 分析表明,这些基因参与modification-dependent macromolecule catabolic process、modification-dependent protein catabolic process、cellular protein catabolic process、protein ubiquitination、ubiquitin-protein ligase activity、actin binding 等生物学过程,KEGG 分析表明参与Ubiquitin mediated proteolysis 信号通路。
2.5 hub gene 表达水平对OC 患者总体生存情况的影响 由于hub gene 在生物学过程中发挥至关重要作用,在相关通路中,其他基因的调控常受到hub gene 影响,因此,检验hub gene 与OC 患者预后转归情况有重要临床意义。利用OncoLnc 评估了所得的274 个hub gene与OC 患者预后相关性的情况,结果发现高表达水平的CDCA3、IQGAP1、BTRC、UBR4、FBXL3、FGF2、SYT1、TRIM4、REPS1、AGFG1、PCNT、POLK、PTGER3和QKI 与OC 患者的总体生存率(OS)降低显著相关(P<0.05);低表达水平的EXO1、MCM3、POLR2D、ANAPC11、SPC24、KLHL25、LSM4、PUF60 和EIF3M与OC 患者的OS 降低显著相关(P<0.05)。
3 讨论
据相关统计表明,大约70%的OC 患者在首次明确诊断时已属肿瘤晚期(III 或IV 期),其5 年生存率<30%;然而,能早期(I 或II 期)明确诊断的患者5年生存率高达70%~90%[13],故开发敏感且可靠的生物标志物以早期诊断OC进而制定有效防治策略具有重要意义。相关研究表明异常表达的lncRNA 与包括OC 在内的恶性肿瘤的发生、耐药及诊断预后密切相关[14-15]。
在本研究中,作者对已发表的有关OC 患者异常表达lncRNA 相关文献进行分析,得到目前已知序列及结构等注释信息的lncRNA 共9 种,随后通过与GSE14407、GSE18520 数据集取交集获得在OC 组织中与上述差异lncRNA 相关的差异表达基因共1,421 个。这些差异基因由478 个上调基因和943 个下调基因组成。这些差异基因在细胞组成(CC)方面主要富集在细胞质、核质、中间体、微管、细胞骨架、细胞膜、有丝分裂核分裂、细胞核和溶酶体膜等部位;在生物学过程(BP)方面主要富集在DNA 复制、细胞分裂、细胞增殖和胞外外泌体等过程;在分子功能(MF)方面主要富集在蛋白质结合过程。KEGG 分析提示49 个差异表达基因(上调18 个,下调31 个)参与了Pathways in cancer 通路。随后,从PPI 网络中筛选出hub gene 274个;经OncoLnc 在线工具分析这些hub gene 与OC 患者的生存预后相关性后发现,14 个基因的高水平表达和9 个基因的低水平表达与OC 患者的不良OS 结局密切相关。
一些hub gene 已在其他实验研究中得到证实与患者的不良预后密切相关。例如,CDCA3 在各种类型癌症的发展中起着关键作用[16-17]。本研究提示,CDCA3在OC 患者中表达水平异常升高,且与OC 患者的不良预后相关(P<0.05),提示CDCA3 有望作为肿瘤预后标志物。又如,在先前报道的OC 研究中,IQGAP1 在OC浸润前期的高表达水平和弥散性表达模式与不良预后显著相关,表明IQGAP1 可能是OC 的潜在预后标志物。就目前诊治水平而言,OC 的预后仍较差,这与OC 患者明确诊断时间较晚及患者并发广泛的腹膜内转移密切相关。本研究中的预后分析表明IQGAP1 与OC 的OS不良预后密切相关。若进一步深入研究其致病性、转移等恶性生物学行为机理,则有望指导IQGAP1 高表达和弥散性表达患者个体化随访频率并设计出更为有效的治疗方法。其他hub gene,如EXO1、POLR2D、BTRC等在本研究中均提示与OC 的不良预后密切关系,但这些基因在肿瘤方面的研究报道甚少,故有进一步研究挖掘的潜在意义。
综上所述,与lncRNA 相关联的hub gene 的异常表达与OC 患者的不良OS 预后密切相关,一些hub gene如MCM3、CDCA3、IQGAP1、KLHL25 及SPC24 等在其他实验研究中也已得到证实,相对较多的hub gene 与肿瘤的预后相关性尚未见于文献报道。目前关于lncRNA及其靶基因协同作用在OC 的基础和临床研究较少,故值得进一步探究,因此,本研究对于开展lncRNA 及与之共表达的相关基因对OC 的诊断、预后等实验生物学研究具有一定的启示作用。下一步,本课题组将对筛选的部分差异基因进行实验和临床双重验证,并将追踪更新的芯片数据进行生物信息学分析。
