超高效液相色谱-串联质谱法检测水产品中10种氨基糖苷类药物残留
2023-05-06李佩佩何鹏飞严忠雍
李佩佩,何鹏飞,严忠雍,方 益
(浙江省海洋水产研究所水产品加工与质量安全研究室/浙江省海洋渔业资源可持续利用技术研究重点实验室,浙江 舟山 316021)
氨基糖苷类化合物(Aminoglycosides,AGs) 虽然有一定的耳毒性、肾毒性和神经肌肉阻滞作用[1,2],但其价格低廉,因此在临床和兽药领域应用广泛,是一类广谱抗菌剂和促生长的饲料添加剂[3,4]。目前多个国家和组织建立了AGs 在动物源性食品中的相关限量标准[5-6]。AGs 分子中因富含氨基和羟基而呈强极性,在反相色谱中的保留性差,分子中缺少发色团和荧光团,在常规检测器上没有响应,因此动物源食品中AGs 的定量检测比其他抗生素更复杂。目前,对AGs 的检测方法主要有免疫分析法[7~9]、毛细管电泳法[10~13]、液相色谱法(LC)[14,15]和液相色谱-串联质谱法(LC-MS/MS)[16~19]等。免疫分析法易出现假阳性,重复性差,更适合初步筛查和半定量分析;毛细管电泳法分离度和检测灵敏度低,分离柱寿命短;液相色谱荧光或者紫外检测法需柱前或柱后衍生,衍生物产量低和稳定性差,导致方法重现性差;HPLC-MS/MS 法由于定性定量准确,灵敏度高、分析种类多等特点而得到广泛应用。动物源食品中AGs 的分析种类从一种发展到最多可同时检测20多种[20]。
当前的研究多集中于牛奶[20~24]、鸡蛋[25,26]、牛鸡羊猪组织[27~28]等畜禽动物源食品以及蜂蜜[29~31],对于水产品中多种氨基糖苷类药物同时检测的研究并不多见[32]。本研究选择壮观霉素、潮霉素B、双氢链霉素、链霉素、丁胺卡那霉素、卡那霉素、安普霉素、妥布霉素、庆大霉素、新霉素10种典型和应用较多的氨基糖苷类药物为代表物质,建立同时检测水产品中10 种氨基糖苷类药物的超高效液相色谱串联质谱方法,为水产品中多种AGs 药物的残留监管提供分析方案。
1 材料与方法
1.1 材料与主要仪器
壮观霉素(Spectinomycin,SPEC,纯度97.3%),潮霉素B(Hygromycin B,HYGRO,纯度93.5%),双氢链霉素(Dihydrostrepmycin,DHSTREP,纯度97.7%),链霉素 (Streptomycin,STREP,纯度89.8%),丁胺卡 那霉素(Amikacin,AMIK,纯度89.7%),卡那霉素 (Kanamycin,KANA,纯度96.1%),安普霉素(Apramycin,APRA,纯度80.4%),妥布霉素(Tobramycin,TOBRA,纯度82.2%),庆大霉素(Gentamycin,GEN,纯度87.4%),新霉素(Neomycin,NEO,纯度76.6%)均购自德国Dr.Ehrenstorfer 公司。磷酸二氢钾、乙二胺四乙酸二钠、三氯乙酸、盐酸、氢氧化钠均为分析纯,购自上海国药集团化试剂有限公司。甲醇、乙腈、甲酸均为色谱纯,购自德国Merck 公司。实验用水为Milli-Q 制备一级水(电阻率≥18.2 MΩ·cm)。
AcquityTM超高效液相色谱仪(UPLC)、Quattro Premier XE 串联四极杆质谱仪(美国Waters 公司);MS3 型旋涡 混合器(德 国IKA 公 司);Centrifuge 5810高速离心机(德国Eppendorf公司);N-EVAP112氮吹仪(美国Organomation 公司);24 通道固相萃取装置(美国Supelco 公司)。Waters Oasis HLB 固相萃取柱(200 mg/6mL,美国Waters 公司)。ME204E/02电子天平(梅特勒-托利多仪器(上海)有限公司);SL502N 电子天平(上海民桥精密科学仪器有限公司)。
1.2 方法
1.2.1 样品制备 从市场上采集鱼、虾、蟹、贝等样品,按照SC/T 3016-2004《水产品抽样方法》处理后于-18 ℃密封保存。使用前先解冻。鱼,去鳞、去皮,沿脊背取肌肉;虾,去头、去壳、去肠腺,取肌肉部分;鳖、蟹和贝类,取可食部分,切成不大于0.5 cm×0.5 cm×0.5 cm的小块,混匀,充分匀浆,于-18 ℃以下冷冻保存,备用。
1.2.2 样品前处理 称取匀质的样品(2.0 ± 0.02 g)于50 mL 离心管,加入10 mL 10 mmol/L 磷酸二氢钾缓冲溶液(磷酸二氢钾1.36 g,乙二胺四乙酸二钠0.15 g,三氯乙酸20.0 g,pH 4.0,定容至1 000 mL),涡旋振荡提取5 min,以7 000 r/min离心5 min,取上清液至50 mL 离心管,残渣用8 mL 10 mmol/L 磷酸二氢钾缓冲溶液重复提取1 次,合并提取液,混匀,用10 和1.0 mol/L NaOH 调pH 值为6.8 ± 0.2,用水定容至20 mL,混匀,用快速滤纸过滤,待净化。
HLB固相萃取柱预先用5 mL甲醇和5 mL水活化。准确吸取10 mL 提取液过柱,待样液完全流出后,依次用5 mL水和5 mL体积分数5%的甲醇溶液淋洗,弃去全部流出液。挤干或者减压抽干柱子,用5 mL 体积分数80%乙腈(含体积分数1%甲酸)洗脱。收集洗脱液于15 mL 离心管中,水浴氮吹浓缩至近干(温度低于45 ℃),用体积分数0.1%甲酸-乙腈(体积比2∶8)溶液定容至1.0 mL,经0.22 μm 有机微孔滤膜过滤后供HPLC-MS/MS分析。
1.2.3 工作溶液配制及标准工作曲线制作 标准储备溶液:称取标准品5.00 mg,用甲醇溶解,定容至50.0 mL,按照各自硫酸盐和纯度计算得到质量浓度约100 μg/mL的标准储备液。
混合标准工作溶液:准确移取适量10种单标贮备液,用水定容至50 mL,配制成壮观霉素、双氢链霉素、链霉素、丁胺卡那霉素、卡那霉素、妥布霉素、庆大霉素分别为4.0 μg/mL,潮霉素B、安普霉素分别为10.0 μg/mL,新霉素为20.0 μg/mL 的混合标准溶液。
基质匹配标准工作曲线:称取6 份空白样品于50 mL 离心管,按照样品前处理方法处理后得到适量的复溶溶液,用0.22 μm 有机微孔滤膜过滤后代替流动相,分别移取适量的混合标准工作溶液,以过滤后的复溶液定容至1.0 mL,以各个浓度为横坐标,各个浓度对应的质谱响应值为纵坐标,制作基质匹配工作曲线。
标准工作曲线:分别移取适量的混合标准工作溶液,用体积分数0.1%甲酸-乙腈(体积比2∶8)溶液定容至1.0 mL,以各个浓度为横坐标,各个浓度对应的质谱响应值为纵坐标,制作标准工作曲线。
在优化的色谱质谱仪器条件下,配制壮观霉素、双氢链霉素、链霉素、丁胺卡那霉素、卡那霉素、妥布霉素、庆大霉素质量浓度为2、5、10、25、50、100 ng/mL,潮霉素B、安普霉素质量浓度为5.0、12.5、25.0、62.5、125.0、50.0 ng/mL,新霉素质量浓度为10、25、50、125、250、500 ng/mL 的基质匹配标准工作溶液系列进UPLC-MS/MS 测定。以定量离子的峰面积对质量浓度进行线性回归,绘制标准曲线。
1.2.4 色谱及质谱条件 色谱条件:SIELC Obelisc R 柱(100 mm ×2.1 mm,5 μm);进样量5 μL;样品室温度10 ℃,柱温40 ℃;流动相A 为体积分数1%甲酸(含2 mmol/L 乙酸铵),B 为乙腈,梯度洗脱时流速为0.3 mL/min,程序如表1所示。
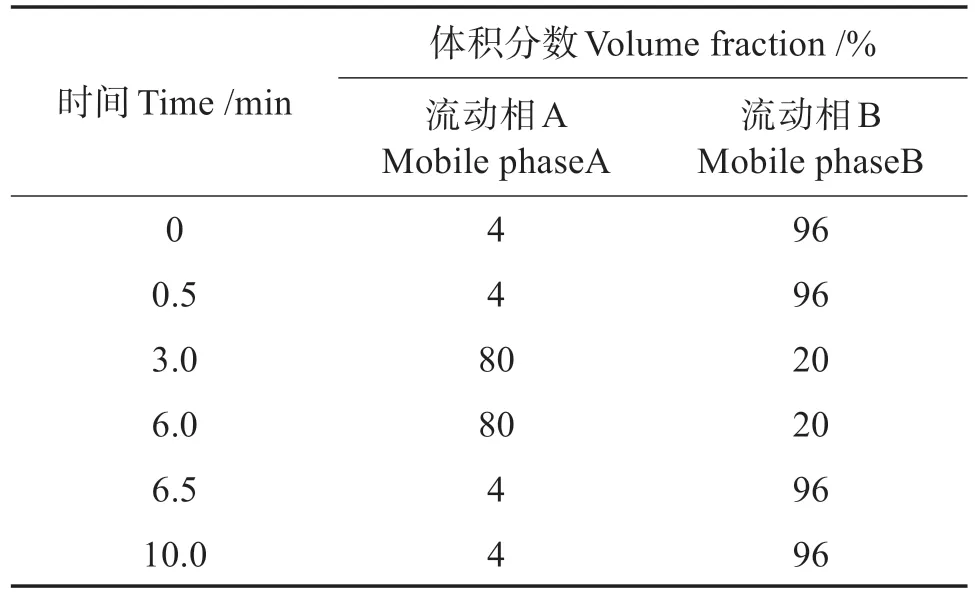
表1 10种氨基糖苷类药物的色谱梯度洗脱程序Table 1 Gradient elution procedures of 10 kinds of AGs
质谱条件:电喷雾离子源;正离子扫描(ESI+)模式扫描;多反应监测(MRM) 方式;毛细管电压:3.5 kV;离子源温度:120 ℃;脱溶剂气温度:380 ℃;锥孔气为高纯氮气,流量:50 L/h;脱溶剂气为高纯氮气,流量:600 L/h。
1.2.5 上样液pH 值的确定 实验考察pH 值不同的上样液中10 种AGs 在HLB 柱上的保留情况,以回收率表示。取4.0 μg/mL 混合标准溶液500 μL 加入到4 mL 10 mmol/L 磷酸二氢钾缓冲溶液中,涡旋混匀提取后,调节溶液pH 值分别为5.0、6.0、6.5、7.0、7.5、8.0、8.5,然后将提取液全部过柱,采用相同淋洗条件淋洗,氮吹复溶过膜后进质谱检测10种AGs含量,计算回收率,每个pH值做3个平行。
1.2.6 洗脱液组成的确定 向10 mL 磷酸二氢钾缓冲溶液中加入一定体积的混合标准溶液,调节pH值为6.8,全部过固相萃取柱,分别用体积分数80%乙腈溶液(含体积分数1%甲酸)、体积分数1%甲酸甲醇、体积分数80%甲醇溶液(含体积分数1%甲酸)等3 种不同洗脱液洗脱,氮吹至近干后用体积分数0.1%甲酸溶液-乙腈(体积比2∶8) 定容至1.0 mL。上机检测比较10 种AGs 回收率,比较3 种洗脱液对10种AG的洗脱效果。
1.2.7 基质效应的计算 分别选择草鱼(Ctenopharyngodon idella)、凡纳滨对虾(Litopenaeus vannamei)、三疣梭子蟹(Portunus trituberculatus)作为空白样品基质,经样品前处理后,得到空白样品基质复溶液,用于配制鱼、虾和蟹类的基质匹配工作溶液。将3种基质匹配标准溶液和试剂标准工作溶液分别进行HPLC-MS/MS 分析,得到各组分的峰面积。以峰面积为纵坐标,标准溶液质量浓度为横坐标绘制标准曲线,采用基质匹配工作曲线和试剂工作曲线的斜率差计算基质效应。
2 结果与讨论
2.1 前处理条件
AGs 化合物结构中含有羟基,有强极性和亲水性,样品宜先用亲水性有机溶剂或酸溶液脱蛋白和提取。三氯乙酸溶液、磷酸二氢钾缓冲溶液、乙酸铵缓冲溶液是3 类常用的提取液[33]。AGs 的提取效率与其分子结构中氨基官能团的数量有某种程度的反相关性,邻近的氨基基团可与多电荷离子形成螯合物而降低提取效率[16]。因此,常需添加离子螯合剂乙二胺四乙酸二钠,以抑制螯合物的形成,提高分析物提取效率。本研究采用含有乙二胺四乙酸二钠的磷酸二氢钾缓冲溶液进行提取。由于提取液中加入三氯乙酸,可有效沉淀蛋白,基本不会造成过柱堵塞现象。
AGs 提取液净化时所用固相萃取柱主要有C18柱,HLB 柱,CBX、SCX、MCX、AmberliteIRC-50 等阳离子交换柱等,也会用专用性更强的分子印迹(MIP) 固相萃取柱[34]。其中HLB 柱更适于多种类AGs 净化,通用性强,结果稳定,且比MIP 的性价比高。本研究采用HLB 柱进行净化,重点考察上样液的pH 值和洗脱液的组成对水产品中10种AGs回收率的影响。
2.1.1 上样液的pH 值 10 种氨基糖苷类分析物分子中分别含有不同数量的氨基和羟基,在不同pH值下每种分析物pKa值有多种,进而影响其在HLB柱上的保留。本研究表明,丁胺卡那霉素、卡那霉素、妥布霉素、庆大霉素、新霉素在所测pH 值范围内回收率变化不大,均可达到在80%以上,其他5种AGs 的变化情况如图1 所示。由图1 可见,5 种AGs在pH 值6.0~7.0 时回收率变化幅度小,略有上升,且回收率均在70%以上。当pH 低于6.0 或大于7.0时,安普霉素、潮霉素和双氢链霉素回收率变化幅度较大。因此,为保证10 种AGs 回收率均达较高,在固相萃取上样前应将提取液的pH 值调节至6.6~7.0。先用10 mol/L NaOH 溶液调节,再用1 mol/L NaOH微调。
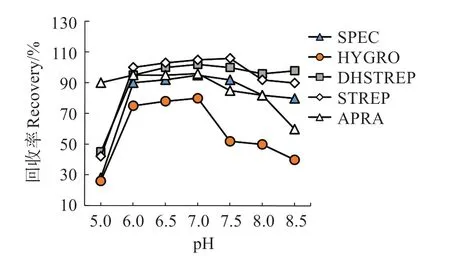
图1 pH对水产品中5种氨基糖苷类药物加标回收率的影响Fig.1 Effect of pH on recoveries of 5 AGs in aquatic products
2.1.2 洗脱液的组成 AGs极性强,洗脱液宜含有一定比例的水溶液,而非纯有机相。结果显示,采用体积分数1%甲酸甲醇洗脱后,壮观霉素,妥布霉素和新霉素的回收率低于50%;体积分数80%甲醇溶液(含体积分数1%甲酸)洗脱后,壮观霉素,妥布霉素和新霉素的回收率有所升高,大于60%,但是10种AGs 的回收率均在80%以下。10 种AGs 在体积分数80%乙腈溶液(含体积分数1%甲酸)洗脱液下回收率均可达80%以上。这与氨基糖苷类药物本身的化学结构和所带基团以及乙腈的洗脱能力等多种因素相关。因此,选择体积分数80%乙腈水溶液(含体积分数1%甲酸)作为洗脱液。
2.2 色谱条件优化
AGs 分子结构中含有氨基糖和氨基环醇,是一类强极性的碱性药物,在常用的C18、C8等反相色谱柱上保留很弱,为实现液相色谱的有效保留和分离,一是在流动相中加入挥发性的七氟丁酸(HFBA)、三氟乙酸(TFA)、五氟丙酸(PFA)等离子对试剂,使其与AGs 结合,形成疏水型离子对,延长AGs 在反相色谱柱上的保留时间;二是直接用可保留AGs 的亲水作用色谱柱(HILIC)。
HILIC 色谱柱的固定相种类很多,包括硅胶、氨基、氰基、氨基甲酸酯、磺酸甜菜碱型两性离子以及其他的极性键合硅胶固定相[34-35]。由于AGs 的极性官能团可在硅胶表面上和硅羟基之间形成氢键或离子交换,传统的裸硅胶柱对极性化合物有强烈的静电吸附作用,从而导致AGs 峰太宽,峰形矮胖,有双峰,不尖锐[36]。不同柱填料的亲水作用色谱对于AGs 的吸附和分离效果有很大影响,两性离子柱Obelisc R,ZIC-HILIC 在AGs 的分离效率和检测灵敏度方面最佳[37-38]。
用C18为色谱柱时,离子对试剂会对质谱离子源和色谱分离系统造成一定损害。为避免使用离子对试剂,选择两性离子柱Obelisc R 为分离柱,其在分离时可形成一“液态分离池”,实现在低浓度缓冲盐条件下有效分离多种AGs。本研究比较体积分数1%甲酸溶液(含2 mmol/L 乙酸铵溶液)-乙腈、体积分数1%甲酸溶液(含2 mmol/L乙酸铵溶液)-体积分数1%甲酸乙腈) 和体积分数1%甲酸溶液-体积分数1%甲酸乙腈对10种AGs的分离效果。结果表明,乙腈中加入甲酸对于分离度无明显提升,而乙酸铵的加入使新霉素和潮霉素B 的峰形变得更为尖锐、对称。通过优化梯度洗脱程序,所有分析物在6 min 内完全出峰,10种分析物分离良好,峰形尖锐,响应值较高。
2.3 质谱条件优化
氨基糖苷类化合物的结构中含有氨基环醇、羟基和伯胺或仲胺基团等,呈强极性和弱碱性,适合电喷雾正离子扫描模式。优化后的质谱条件见表2,10种AGs的MRM图见图2。
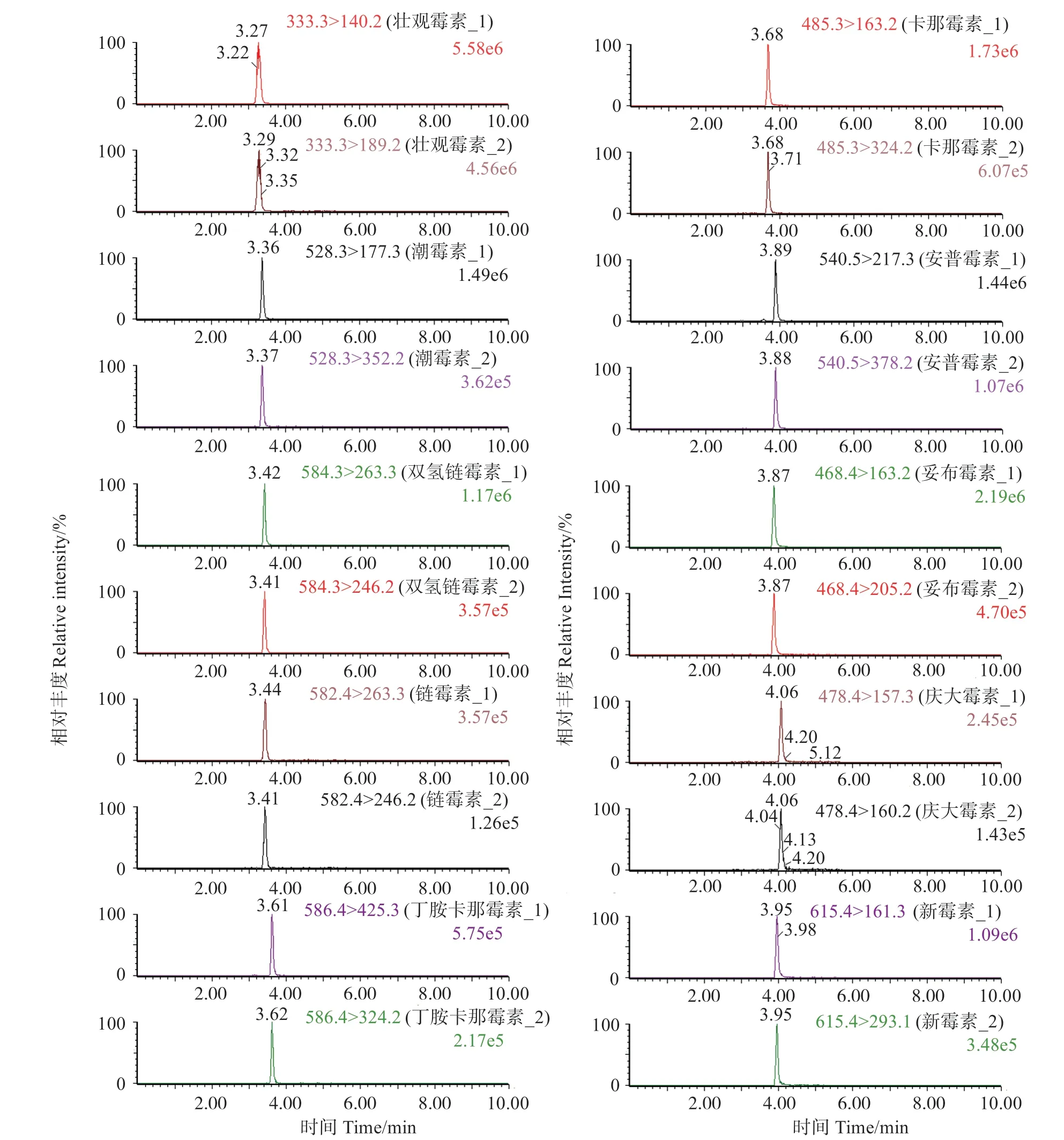
图2 质量浓度为25.0 ng/mL 的10种AGs混合标准溶液的MRM图Fig.2 MRM chromatograms of 10 kinds of AGs mixed standard solution with mass concentration of 25.0 ng/mL
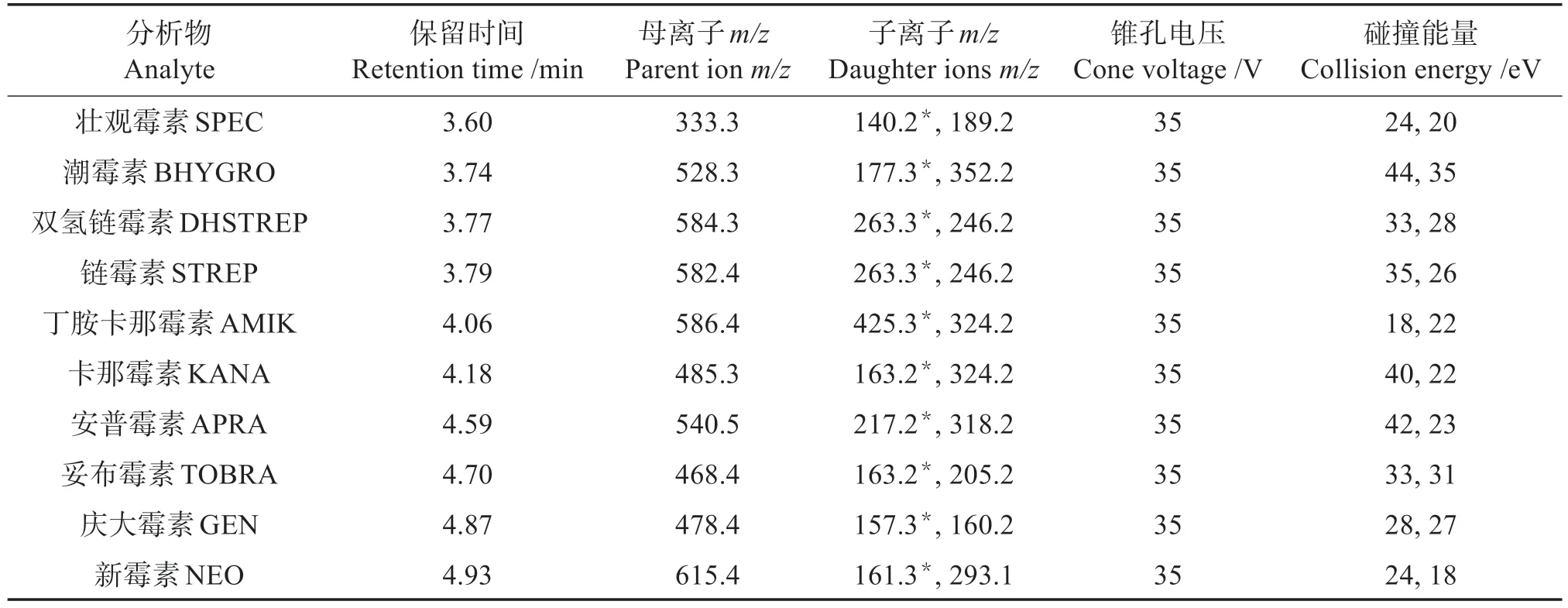
表2 10种氨基糖苷类药物的多反应监测实验条件Table 2 Conditions of multiple reaction monitoring for 10 kinds of AGs
2.4 基质效应
氨基糖苷类化合物质谱检测时有较大的基质效应,有必要考察10种AGs在不同种类水产品中的基质效应。图3 表明,在草鱼、凡纳滨对虾中,10 种AGs 的基质效应较为一致,虾的基质效应略低于鱼;三疣梭子蟹的基质效应低于鱼和虾。壮观霉素、潮霉素B、卡那霉素的基质效应较高。因此,水产品中AGs 含量测定时应制作空白基质标准曲线以抵消基质效应,且不同种类的水产品需分别制作对应空白基质曲线,以更准确定量。
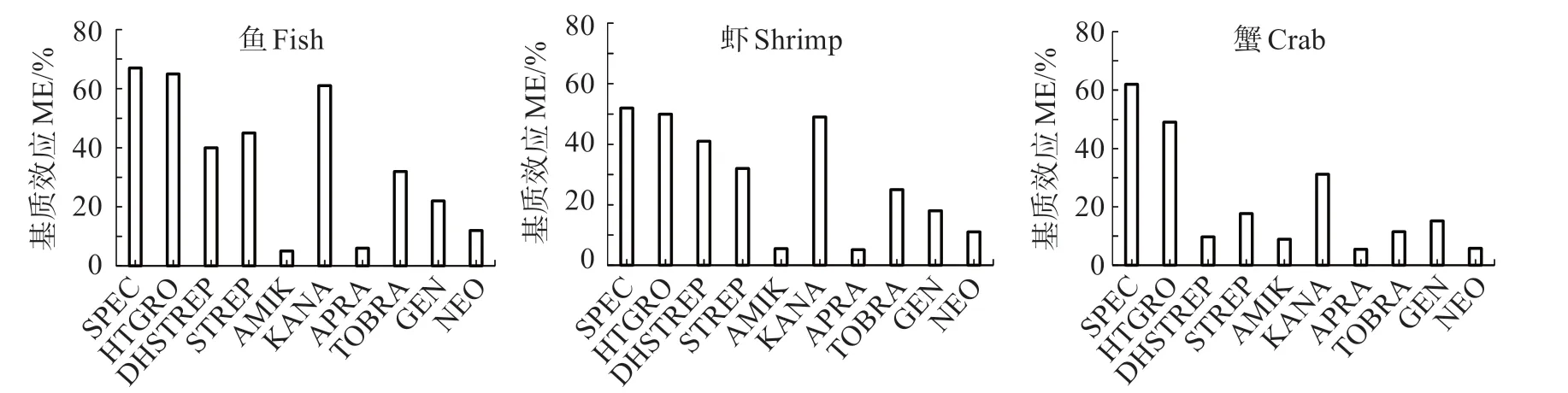
图3 三类水产品中10种AGs药物的基质效应评价结果Fig.3 Matrix effect evaluation results of 10 AGs drugs in three types of aquatic product
2.5 线性范围、检出限和定量限
表3表明,壮观霉素、双氢链霉素、链霉素、丁胺卡那霉素、卡那霉素、妥布霉素、庆大霉素在2.0~100 ng/mL,潮霉素B、安普霉素在5.0~250 ng/mL,新霉素在10~500 ng/mL 范围内相关系数均大于0.999,满足仪器分析的需要。以信噪比(S/N)≥3 确定的检出限,以S/N≥10 确定的定量限(Limit of quantification,LOQ)见表3。
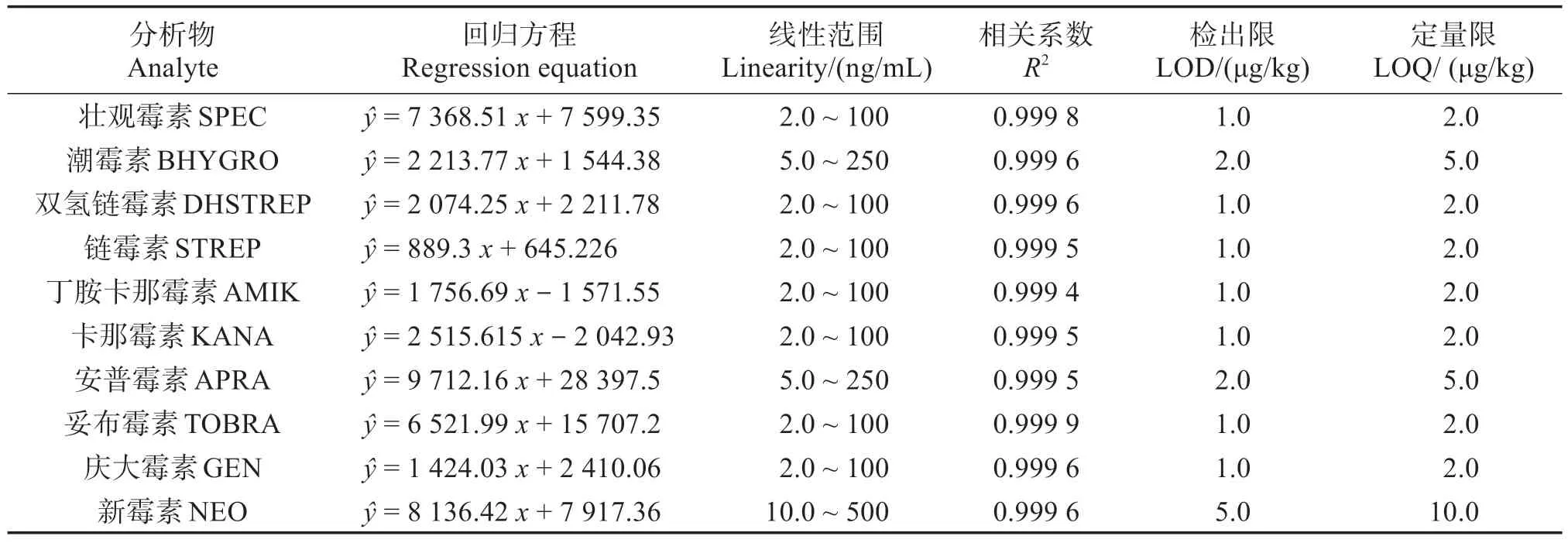
表3 10种AGs的回归方程、线性范围、方法检出限和定量限Table 3 Calibration equation,linear range,limit of detection(LOD)and limit of quantification(LOQ)for 10 kinds of AGs
2.6 方法回收率和精密度
为考察方法的准确度和精密度,以草鱼、凡纳滨对虾和三疣梭子蟹样品为空白基质进行添加回收实验,表4 表明,10 种AGs 药物的平均回收率为74.8%~104.5%,相对标准偏差(RSD)为4.5%~12.6%,方法的精密度和准确度可满足分析检测要求。
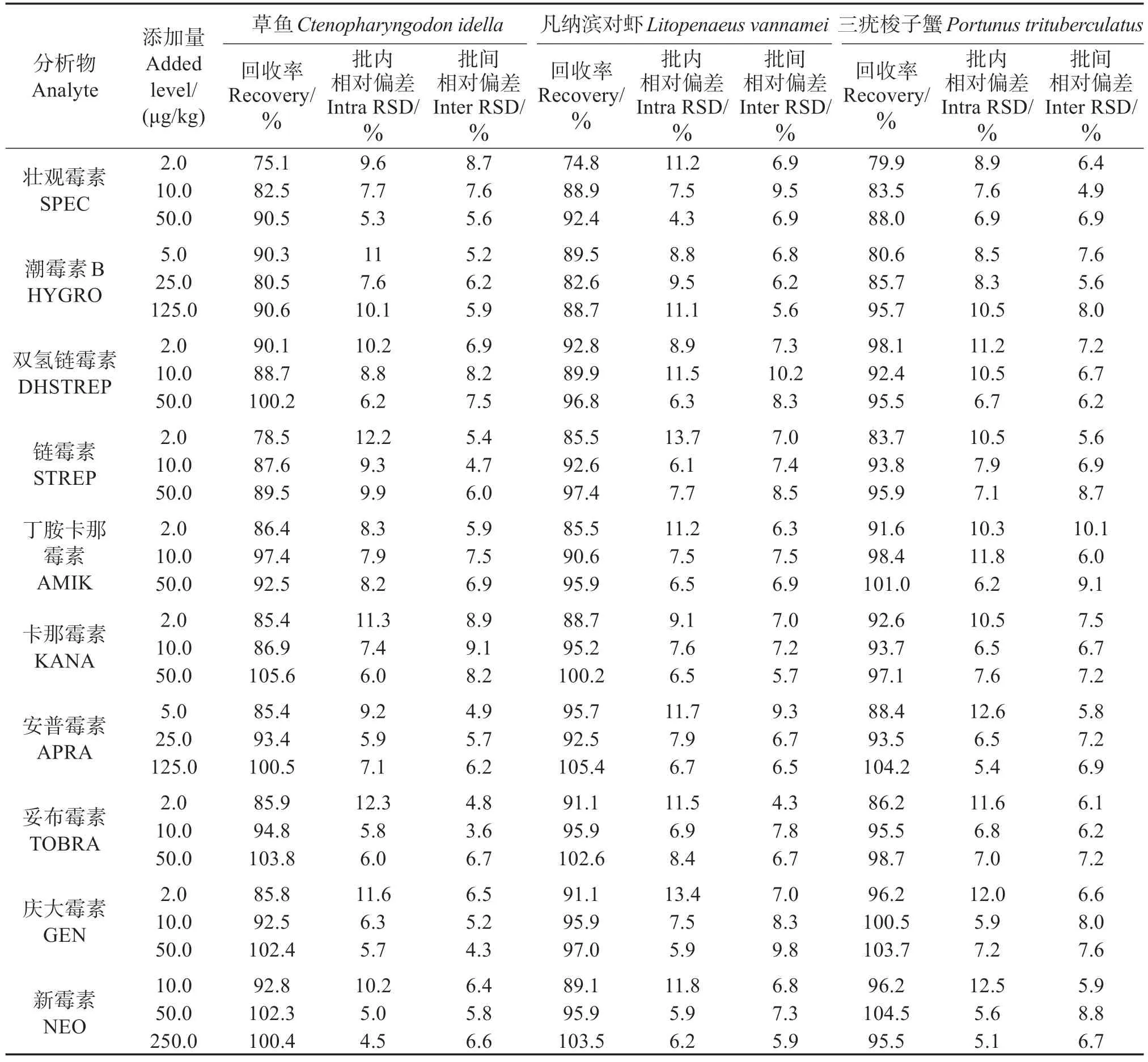
表4 空白试样添加回收率和精密度测定结果(n=6)Table 4 Results of recovery and precision of spiked samples
2.7 实际样品检测
为考察方法的适用性和实用性,选取2022 年农业农村部水产品质量安全城市例行监测和2022 年舟山市初级水产品质量安全专项监测任务的部分代表性样品70 份,包括草鱼(C.idella)、大黄鱼Larimichthys crocea、黑鲷(Acanthopagrus schlegelii)、乌鳢(Channa argus)、鳜(Siniperca)、凡纳滨对虾(L.vannameiBoone)、哈氏仿对虾(Parapenaeopsis hardwickii)、中华管鞭虾(Solenocera melantho)、梭子蟹(Portunus trituberculatus)等,采用本研究建立的方法测定10 种AGs 药物的残留量。检测结果显示以上水产品中不含10 种AGs 类药物。
3 结论
本研究建立了超高效液相色谱-串联质谱同时测定水产品中10 种AGs 药物的方法。通过选择合适的固相萃取上样液pH 和合适的洗脱液,可确保分析物的有效提取和富集,10种AGs药物的平均回收率为74.8%~104.5%,相对标准偏差为4.5%~12.6%;通过选择适宜的梯度洗脱条件,联合色谱的高效分离和串联质谱的高选择性,成功实现10种物质的快速有效分离和准确定量。方法灵敏度高,回收率和精密度良好,适用于水产品中10种AGs药物的同时测定。
