LPL基因变异导致的婴儿家族性高乳糜微粒血症合并肾脏错构瘤1例报告
2023-04-29陈欣涛方微园龚晓妍林琼陆怡
陈欣涛 方微园 龚晓妍 林琼 陆怡
关键词:脂蛋白脂酶缺乏症Ⅰ型; 高甘油三酯血症; 肾脏错构瘤
基金项目:国家重点研发计划(2021YFC 2700800)
Infantile familial chylomicronemia syndrome caused by LPL gene variants coexisting with renal hamartoma: A case report
CHEN Xintao1a,2, FANG Weiyuan1a, GONG Xiaoyan1b, LIN Qiong2, LU Yi1a. (1. a.The Center for Pediatric Liver Diseases, b. Department of Clinical Nutrition, Childrens Hospital of Fudan University, Shanghai 201102, China; 2. Department of Gastroenterology, Wuxi Childrens Hospital, Wuxi, Jiangsu 214023, China)
Corresponding author:
LU Yi, luyi@fudan.edu.cn (ORCID:0000-0002-3311-4501)
Key words:
Hyperlipoproteinemia Type I; Hypertriglyceridemia; Renal Hamartoma
Research funding:National Key Research and Development Program of China(2021YFC 2700800)
1 病例资料
患儿男性,4月龄+20天,因“发现血脂异常8天”于2021年3月收治复旦大学附属儿科医院肝病科。患儿8天前因“发热、咳嗽半天”于当地医院就诊,生化检查发现血浆甘油三酯(TG)78.3 mmol/L(0~1.7 mmol/L),总胆固醇(TC) 9.0 mmol/L(0~5.18 mmol/L),病因未明。患儿既往无反复哭闹、恶心呕吐、腹胀、皮肤黄色瘤等表现。患儿为第2胎第2产,疤痕子宫足月剖宫产娩出,出生体质量3.25 kg,生后无缺氧窒息史,母乳喂养,尚未添加辅食;生长发育同同龄正常婴儿。第1胎第1产为3岁哥哥,体健;父母体健;非近亲结婚,血脂水平及肝功能均未见异常;无脂肪肝、胰腺炎、冠心病等家族史。入院时查体:身长65 cm,体质量8.5 kg,无特殊面容,腹部稍膨隆,肝肋下2 cm,剑突下1 cm,质地软,脾肋下 4 cm,质地中等,四肢无畸形,肌力肌张力未见异常。
辅助检查:空腹3 h抽取静脉血静置后试管上层可见显著乳白色分层,查生化示TG 126.5 mmol/L,TC 10.2 mmol/L,高密度脂蛋白0.12 mmol/L(1.03~1.55 mmol/L),低密度脂蛋白5.85 mmol/L(1.89~4.21 mmol/L),游离脂肪酸 631 μmol/L(172~586 μmol/L),脂蛋白a 30.6 mg/dL(0~30 mg/dL),载脂蛋白A1 0.54 g/L(1.0~1.6 g/L),载脂蛋白B 1.15 g/L(0.1~1.1 g/L),淀粉酶35 U/L(40~132 U/L),脂肪酶27 U/L(0~60 U/L),葡萄糖4.2 mmol/L(3.9~6.1 mmol/L);肝功能、肾功能、肌酶、电解质、血乳酸、血尿串联质谱、甲状腺功能、皮质醇均未见明显异常。
腹部超声检查示:脾脏肿大伴双肾多发实质性占位。腹部增强磁共振示:肝脏形态可,肝内未见明显异常信号影。双肾内见多发小圆形T2WI高低混杂信号、T1WI等低信号影,增强后呈相对低信号,直径3.0~4.8 mm,T2WI高信号影抑脂后信号明显减低;另右肾见斑片状T2WI混杂信号灶,横截面约14.1 mm×9.4 mm,增强后强化不明显;考虑双肾错构瘤可能性大。心超、心电图均未见显著异常。眼科会诊眼底未见异常。
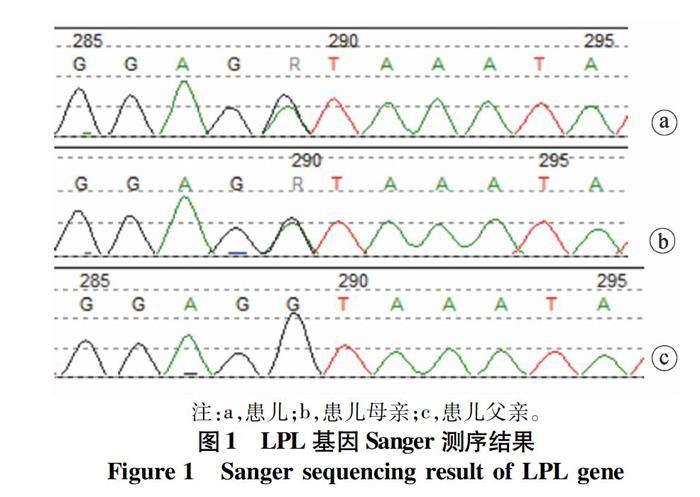
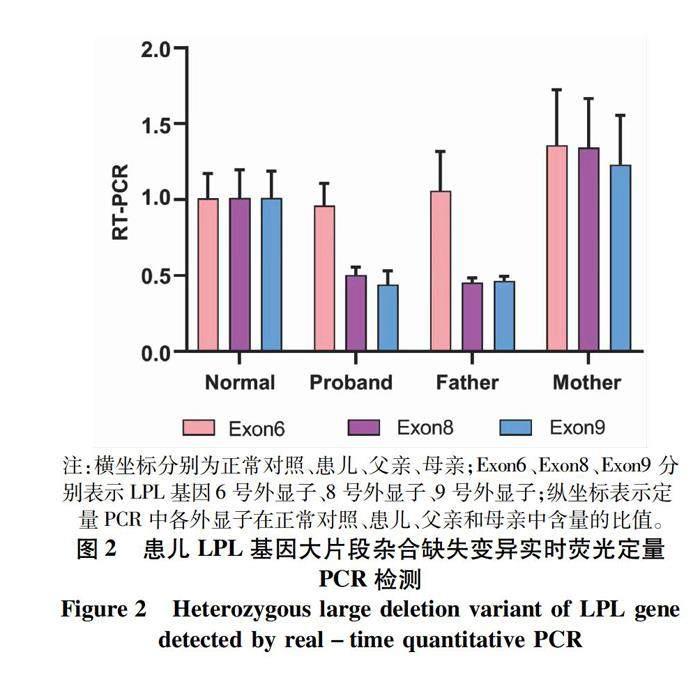
全外显子基因组测序结果提示LPL基因(NM_000237)5号外显子存在经典剪切变异c.775+1G>A(图1),来源于母亲,还提示存在父源LPL基因大片段杂合缺失。再次征得监护人知情同意后对其一家三口血液标本行实时荧光定量PCR檢测,结果示患儿及父亲存在LPL基因8号及9号外显子缺失(图2)。
2个变异均未在HGMD数据库中报道,在ESP数据库、千人数据库、EXAC数据库正常对照人群中亦未见报道。根据美国医学遗传学与基因组学学会基因变异解读指南:c.775+1G>A (PVS1+PM2+PM3+PP4),外显子8/9缺失(PVS1+PM2+PM3+PP4),2个变异评级均为致病性变异(基因测序方法及解读方法见文献[1-2])。患儿2个LPL基因新发变异与TG极重度升高的临床表现相符合,是导致其高甘油三酯血症的遗传基础。
肝病科及营养科医生协同治疗该患儿,予以低脂饮食,先后予以脱脂奶粉,去脂肪母乳,富中链脂肪酸配方奶,去脂米粉,低脂纯瘦荤菜辅食、中链脂肪酸为烹饪食用油等低脂方式喂养,并补充脂溶性维生素;期间监测TG水平波动于5.4~24.3 mmol/L,且发现其水平受饮食影响大,嘱家属依从性加强,后随访多次TG水平波动于1.8~4.3 mmol/L,生长运动发育佳,同正常同龄儿童。双肾多发实质性占位予以随访,多次复查腹部超声,占位大小和性质同前,无进一步进展和恶化。
2 讨论
LPL基因位于第8染色体p22,长度约30 kb,包含10个外显子和9个内含子,编码包含475个氨基酸的蛋白质,即脂蛋白脂肪酶(lipoprotein lipase,LPL)。LPL是脂肪酶家族中一员,主要在脂肪组织、心肌和骨骼肌实质细胞粗面内质网合成,再被分泌、转运到
血管内皮细胞腔面,与血液循环中富含TG脂蛋白(如乳糜微粒和极低密度脂蛋白)直接接触,将其包含的TG分解产生游离脂肪酸,为各个组织供能,是脂质代谢
途径中的关键酶[3-4]。当LPL基因或与LPL功能相关的其他基因(APOC2、APOA5、LMF1和GPIHBP1)[5-8]发生变异时均可导致LPL缺乏或功能受损,使血循环中TG水解受阻,引起乳糜微粒大量堆积,血浆TG水平显著升高(880 mg/dL或10 mmol/L)[9],引发的一系列临床症状被称为家族性高乳糜微粒血症(familial chylomicronemia syndrome,FCS)或脂蛋白脂肪酶缺乏症,又称Ⅰ型高脂蛋白血症(OMIM 238600)[7,10]。其中,LPL基因变异占FCS病因90%以上,发病率约为1/1 000 000[11],男女发病比率无明显差异,可在任何年龄出现症状,但典型FCS常在儿童早期或青年期就出现临床症状[7,11]。临床表现包括反复发作的腹痛、胰腺炎、皮肤黄瘤、肝脾肿大和视网膜脂血症,严重程度与乳糜微粒血症严重程度相关,后者又与膳食中脂肪摄入量密切相关。反复发作性急性胰腺炎是FCS最严重的并发症[11],和其他胰腺炎相比,FCS因严重高甘油三酯血症诱发的急性胰腺炎病情更严重、多器官衰竭、胰腺坏死和死亡的发生率更高[12]。
FCS血脂异常特点为血TG严重升高,TC升高,低密度脂蛋白和极低密度脂蛋白正常或降低,部分病例报道低密度脂蛋白也可轻度升高;FCS可根据临床表现、血TG显著升高,结合LPL活性或基因分析结果确诊。2017年欧洲临床专家小组[13]提出FCS评分法用于FCS识别和诊断。本病例为婴儿起病,TG极重度升高,多次复查空腹TG仍高,低脂饮食后TG可下降,伴脾肿大,无肥胖、糖尿病、甲状腺功能减退、肾脏疾病、肝脏疾病、药物等继发性高甘油三酯血症高危因素,故首先考虑原发性高甘油三酯血症可能性大。原发性高甘油三酯血症(也称遗传性或家族性高甘油三酯血症)通常由基因缺陷所致,多具有家族聚集性,呈常染色体隐性(autosomal recessive inheritance, AR)或显性遗传(autosomal dominant inheritance,AD),包括FCS(AR)、家族性高甘油三酯血症(AD)、家族性混合型血脂异常(多基因AD)、家族性异常β脂蛋白血症(AD/AR)[14]等。该患儿TG极重度升高,TC轻度升高,低密度脂蛋白胆固醇轻度升高或正常范围,高密度脂蛋白胆固醇水平降低,且父母血脂水平均未见异常,故首先考虑FCS可能性大,全外显子基因测序证实为LPL基因复合杂合变异导致的FCS。病程中患儿皮肤未见黄色瘤,眼底未见视网膜脂血症,无急性胰腺炎等依据,考虑与确诊时年龄尚小、临床表现尚未呈现有关,对于其已知的远期并发症,仍需积极预防,需密切监测和随访。
患儿腹部磁共振检查提示肾脏多发占位,考虑错构瘤可能性大。肾错构瘤又称为肾血管平滑肌脂肪瘤,是由异常增生的血管平滑肌及脂肪组织按不同比例构成的一种良性肿瘤[15]。目前认为是一种良性遗传性疾病,发病原因可能与肿瘤样本中X 染色体失活、染色体异常以及基因变异或基因杂合性缺失有关[16],大约有50%肾错构瘤发生在结节性硬化患者中,而后者由TSC1或TSC2基因变异所致[17-18]。该患儿全外显子基因组测序未发现TSC1/TSC2基因致病性变异。在英文数据库PubMed/Google Scholar和中文万方/知网数据库中搜索LPL基因变异所致FCS病例,复习相关文献未发现FCS合并肾错构瘤或其他肾脏肿瘤的报道,因此该患儿肾脏占位(肾错构瘤)和其已确诊的LPL基因复合杂合变异之间的相关性尚不能建立。目前随访患儿肾脏占位(肾错构瘤)未见增大、出血等。
迄今为止已报道220多个LPL基因变异位点,多集中在外显子5和6[19],占所有变异74%;变异类型中约70%是错义变异,其他还包括无义变异、移码变异、剪切位点变异以及单个外显子或多个外显子的缺失、复制、插入或复杂重排等变异。本例患儿LPL基因上存在分别来自父母的经典剪切变异c.775+1G>A和外显子8/9缺失,均为严重的功能缺失性变异,从分子水平确定了这2个变异为该患儿高甘油三酯血症的遗传学病因;同时这2个变异均为新发变异,进一步丰富了LPL基因变异谱,为家系咨询和产前诊断提供了依据。
胰腺炎是FCS的严重并发症,FCS治疗关键是降低TG使其尽可能接近人体正常水平,从而预防胰腺炎等并发症的发生,提高生活质量;通常建议将血浆TG浓度降低至5.6~10 mmol/L(500~1000 mg/dL)以下[7,20]。治疗方法主要为低脂饮食,成人FSC建议摄入脂肪量小于总能量的15%,儿童则不建议將膳食脂肪摄入量降低至20%以下[12]。此外还要注意避免服用可能会导致TG升高的药物,如糖皮质激素、雌激素、利尿剂等。基因治疗曾于2014年上市,但在2017年因经济因素已停止销售[21]。截至目前国内尚未有儿童高甘油三酯血症治疗的指南或共识,尚无明确的儿童高TG治疗适应证,也尚未有18岁以下已批准使用的降TG药物[22-23];成人降TG常用药物如贝特类药物、omega-3脂肪酸、他汀类药物、烟酸、奥利司他、胰岛素敏感疗法等均提示对FCS治疗效果有限、证据不足或有副作用[7,24];而新型降TG药物微粒体甘油三酯转移蛋白抑制剂可能会导致肝脂肪变性和肝硬化;二酰基甘油酰基转移酶1抑制剂已在成人FCS临床试验中显示有降TG作用,但腹泻发生率较高[12,25];载脂蛋白C-Ⅲ反义寡核苷酸(antisense oligonucleotide,ASO)药物volanesorsen在成人FCS Ⅲ期临床试验中可显著降低TG水平,2019年已在欧洲获批上市,可用于对饮食控制和降TG疗法效果不佳、有急性胰腺炎高风险且经基因检测证实的成人FCS,但有血小板减少风险;新型药物血管生成素样蛋白3抗体及其ASO也可降低TG水平,但尚未在FCS中行临床试验。因此对于儿童FCS目前仍以低脂饮食最为关键,并需注意补充必需脂肪酸、脂溶性维生素及矿物质等;中链脂肪酸可以绕过乳糜微粒形成直接被门静脉吸收至肝脏提供能量,婴儿期可予含中链脂肪酸配方奶,儿童期可用富含中链脂肪酸的食用油烹饪。本病例患儿通过低脂饮食治疗后TG可降至1.8~4.3 mmol/L,控制满意,未加用降脂药物。
总之,临床医师对于儿童期不明原因的重度高甘油三酯血症、肝脾大、胰腺炎等表现的患儿需考虑到FCS可能。早期诊断、及时饮食干预,可减少其并发症尤其是急性胰腺炎发生的风险。基因检测可帮助明确诊断。该重度高甘油三酯血症婴儿通过基因检测技术发现未报道的LPL基因变异位点,丰富了LPL基因变异谱,及时且正确的饮食指导让TG水平由极重度升高逐渐恢复至理想水平。但该患儿同时合并双肾错构瘤,既往文献尚未报道LPL基因变异所致FCS和错构瘤之间的相关性,可待进一步的随访和更多病例进一步明确。
伦理学声明:本例报告通过复旦大学附属儿科医院伦理委員会批准,批号:复儿伦审(2020)402号,已获得患者家属知情同意。
利益冲突声明:所有作者均声明不存在利益冲突。
作者贡献声明:陈欣涛、方微园负责文章的构思与设计,数据收集整理,资料分析,撰写论文,贡献相当,排名不分先后;龚晓妍参与数据分析和解释;林琼、陆怡负责拟定写作思路,指导撰写文章,质量控制及审校。
参考文献:
[1]YANG L, DONG XR, PENG XM, et al.Evaluation of turn around time and diagnostic accuracy of the next generation sequencing data analysis pipeline version 2 of Childrens Hospital of Fudan University[J]. Chin J Evid Based Pediatr, 2018, 13(2): 118-123. DOI: 10.3969/j.issn.1673-5501.2018.02.008.
杨琳, 董欣然, 彭小敏, 等. 复旦大学附属儿科医院高通量测序数据分析流程(第二版)对遗传疾病候选变异基因筛选用时和准确性分析[J]. 中国循证儿科杂志, 2018, 13(2): 118-123. DOI: 10.3969/j.issn.1673-5501.2018.02.008
[2]QIN Q, LIU B, YANG L, et al. Application of copy number variation screening analysis process based on high-throughput sequencing technology[J]. Chin J Evid Based Pediatr, 2018, 13(4): 275-279. DOI: 10.3969/j.issn.1673-5501.2018.04.007.
秦谦, 刘博, 杨琳, 等. 基于高通量测序技术的拷贝数变异筛选分析流程的建立及应用[J]. 中国循证儿科杂志, 2018, 13(4): 275-279. DOI: 10.3969/j.issn.1673-5501.2018.04.007.
[3]KERSTEN S. Physiological regulation of lipoprotein lipase[J]. Biochim Biophys Acta, 2014, 1841(7): 919-933. DOI: 10.1016/j.bbalip.2014.03.013.
[4]OLIVECRONA G. Role of lipoprotein lipase in lipid metabolism[J]. Curr Opin Lipidol, 2016, 27(3): 233-241. DOI: 10.1097/MOL.0000000000000297.
[5]BRAHM A, HEGELE RA. Hypertriglyceridemia[J]. Nutrients, 2013, 5(3): 981-1001. DOI: 10.3390/nu5030981.
[6]DRON JS, HEGELE RA. Genetics of hypertriglyceridemia[J]. Front Endocrinol (Lausanne), 2020, 11: 455. DOI: 10.3389/fendo.2020.00455.
[7]FALKO JM. Familial chylomicronemia syndrome: A clinical guide for endocrinologists[J]. Endocr Pract, 2018, 24(8): 756-763. DOI: 10.4158/EP-2018-0157.
[8]TAKAGI A, IKEDA Y. Genetic diagnosis on hypertriglyceridemia-analysis for LPL gene mutations[J]. Nihon Rinsho, 2013, 71(9): 1569-1576.
[9]GARG A, GARG V, HEGELE RA, et al. Practical definitions of severe versus familial hypercholesterolaemia and hypertriglyceridaemia for adult clinical practice[J]. Lancet Diabetes Endocrinol, 2019, 7(11): 880-886. DOI: 10.1016/S2213-8587(19)30156-1.
[10]LIU LX, WU YT. Clinical advances in hypertriglyceridemia[J]. Chin J Evidence-Bases Cardiovascular Med, 2020, 12(4): 504-508. DOI: 10.3969/j.issn.1674-4055.2020.04.34.
刘立新, 武云涛. 高甘油三酯血症临床新进展[J]. 中国循证心血管医学杂志, 2020, 12(4): 504-508. DOI: 10.3969/j.issn.1674-4055.2020.04.34.
[11]CHYZHYK V, BROWN AS. Familial chylomicronemia syndrome: A rare but devastating autosomal recessive disorder characterized by refractory hypertriglyceridemia and recurrent pancreatitis[J]. Trends Cardiovasc Med, 2020, 30(2): 80-85. DOI: 10.1016/j.tcm.2019.03.001.
[12]GALLO A, BLIARD S, DERASMO L, et al. Familial chylomicronemia syndrome (FCS): recent data on diagnosis and treatment[J]. Curr Atheroscler Rep, 2020, 22(11): 63. DOI: 10.1007/s11883-020-00885-1.
[13]MOULIN P, DUFOUR R, AVERNA M, et al. Identification and diagnosis of patients with familial chylomicronaemia syndrome (FCS): Expert panel recommendations and proposal of an “FCS score”[J]. Atherosclerosis, 2018, 275: 265-272. DOI: 10.1016/j.atherosclerosis.2018.06.814.
[14]VALAIYAPATHI B, SUNIL B, ASHRAF AP. Approach to hypertriglyceridemia in the pediatric population[J]. Pediatr Rev, 2017, 38(9): 424-434. DOI: 10.1542/pir.2016-0138.
[15]CALI A, BRUNELLI M, SEGALA D, et al. Angiomyolipoma of the kidney: from simple hamartoma to complex tumour[J]. Pathology, 2021, 53(1): 129-140. DOI: 10.1016/j.pathol.2020.08.008.
[16]CHENG L, GU J, EBLE JN, et al. Molecular genetic evidence for different clonal origin of components of human renal angiomyolipomas[J]. Am J Surg Pathol, 2001, 25(10): 1231-1236. DOI: 10.1097/00000478-200110000-00002.
[17]PFIRMANN P, COMBE C, RIGOTHIER C. Tuberous sclerosis complex: A review[J]. Rev Med Interne, 2021, 42(10): 714-721. DOI: 10.1016/j.revmed.2021.03.003.
[18]CURATOLO P, BOMBARDIERI R, JOZWIAK S. Tuberous sclerosis[J]. Lancet, 2008, 372(9639): 657-668. DOI: 10.1016/S0140-6736(08)61279-9.
[19]GILBERT B, ROUIS M, GRIGLIO S, et al. Lipoprotein lipase (LPL) deficiency: a new patient homozygote for the preponderant mutation Gly188Glu in the human LPL gene and review of reported mutations: 75% are clustered in exons 5 and 6[J]. Ann Genet, 2001, 44(1): 25-32. DOI: 10.1016/s0003-3995(01)01037-1.
[20]OKAZAKI H, GOTODA T, OGURA M, et al. Current diagnosis and management of primary chylomicronemia[J]. J Atheroscler Thromb, 2021, 28(9): 883-904. DOI: 10.5551/jat.RV17054.
[21]STEINHAGEN-THIESSEN E, STROES E, SORAN H, et al. The role of registries in rare genetic lipid disorders: Review and introduction of the first global registry in lipoprotein lipase deficiency[J]. Atherosclerosis, 2017, 262: 146-153. DOI: 10.1016/j.atherosclerosis.2016.08.023.
[22]WITZTUM JL, GAUDET D, FREEDMAN SD, et al. Volanesorsen and triglyceride levels in familial chylomicronemia syndrome[J]. N Engl J Med, 2019, 381(6): 531-542. DOI: 10.1056/NEJMoa1715944.
[23]ELKINS C, FRUH S, JONES L, et al. Clinical practice recommendations for pediatric dyslipidemia[J]. J Pediatr Health Care, 2019, 33(4): 494-504. DOI: 10.1016/j.pedhc.2019.02.009.
[24]CHAUDHRY R, VILJOEN A, WIERZBICKI AS. Pharmacological treatment options for severe hypertriglyceridemia and familial chylomicronemia syndrome[J]. Expert Rev Clin Pharmacol, 2018, 11(6): 589-598. DOI: 10.1080/17512433.2018.1480368.
[25]MEYERS CD, TREMBLAY K, AMER A, et al. Effect of the DGAT1 inhibitor pradigastat on triglyceride and apoB48 levels in patients with familial chylomicronemia syndrome[J]. Lipids Health Dis, 2015, 14: 8. DOI: 10.1186/s12944-015-0006-5.
收稿日期:
2022-07-30;錄用日期:2022-09-13
本文编辑:林姣
