吸电子基团调控苯基喹啉类铱配合物的合成及光物理性能
2023-02-27常桥稳陈祝安晏彩先刘伟平白福全
常桥稳 陈祝安 冯 璐 姜 雯 晏彩先*, 刘伟平 白福全*,
(1昆明贵金属研究所,昆明贵研新材料科技有限公司,稀贵金属综合利用新技术国家重点实验室,云南贵金属实验室有限公司,昆明 650106)
(2吉林大学化学学院,理论化学研究所,长春 130023)
近20年来,铱磷光配合物由于表现出高的发光效率、良好的热稳定性和发光颜色容易调控等优势,在有机发光二极管(OLED)、电化学发光池(LEC)、光催化反应和传感器等领域有着重要的应用,引起了越来越多科研工作者的极大兴趣[1‑10]。苯基喹啉主配体及衍生物已经广泛应用到铱磷光配合物的研究中[11‑17]。大量研究表明:苯基喹啉类铱磷光配合物的HOMO能级主要分布在主配体的苯基和铱(Ⅲ)中心上,LUMO能级主要局域在喹啉环和辅助配体上。在苯基上引入吸电子基团,能降低HOMO能级,增大HOMO‑LUMO能级差,发射波长蓝移;在苯基上引入给电子基团,能提高HOMO能级,减小HOMO‑LUMO能级差,使发射波长红移。Kim等[14]合成了系列甲基取代的苯基喹啉类铱磷光配合物,甲基引入到苯基喹啉中苯基的3位和5位,使铱磷光配合物的发射波长发生了红移,并显著提高了铱磷光配合物的发光效率和外量子产率。陶鹏等[12‑13]合成大量取代苯基喹啉类铱磷光配合物,与(mpq)2Ir(acac)铱磷光配合物相比较,在苯基‑4‑甲基喹啉主配体中苯基的间位和对位上引入甲氧基。当甲氧基的位置处于间位时,配合物的发射波长蓝移17 nm,而当甲氧基的位置处于对位时,配合物发射波长则红移39 nm,甲氧基位于间位和对位的2种不同的配合物,最大发射波长相差62 nm。另外,在苯环上引入不同数量的氟原子,发光颜色从橙红光至红光变化,改变氟的数量实现了铱磷光配合物在560~590 nm范围内调控。由此可见,通过改变取代基的供/吸电子能力、取代点位和取代基的数量均能实现铱磷光配合物发光颜色和发射波长的调控。
取代基的供/吸电子能力又与取代基的取代点位息息相关。通常认为甲氧基是供电子基团,但实际上,甲氧基具有2种相反的电子效应:供电子效应和吸电子效应,这主要与甲氧基所处苯环的位置有关[17]。甲氧基作为吸电子基团在苯基喹唑啉和苯基呋喃吡啶铱磷光配合物中也有报道[18‑20]。我们最近也证实了在苯基喹啉中苯基的2位和4位,甲氧基为吸电子基团[21]。为了进一步探究吸电子基团对铱磷光配合物发光颜色和发射波长的影响,我们用吸电子基团修饰苯基喹啉主配体,分别在苯基空间位阻较小的2位和4位引入吸电子的氟、甲氧基和三氟甲基,设计、合成出3个铱磷光配合物(2,4‑2R‑mpq)2Ir(tmd)(R=F(1)、MeO(2)、CF3(3),2,4‑2R‑mpq=2,4‑二取代基苯基‑4‑甲基喹啉,tmd=2,2,6,6‑四甲基庚二酮),并对其光物理性能进行了研究。
1 实验部分
1.1 试 剂
四(2,4‑二(甲氧基)苯基‑4‑甲基喹啉)二氯化铱二聚体、四(2,4‑二氟苯基‑4‑甲基喹啉)二氯化铱二聚体和四(2,4‑二(三氟甲基)苯基‑4‑甲基喹啉)二氯化铱二聚体均由昆明贵金属研究所自制。其余试剂包括tmd(98%,网化商城)、无水碳酸钠(AR,四川西陇化工有限公司)、乙二醇单乙醚(AR,天津市光复精细化工研究所)、二氯甲烷(AR,四川西陇化工有限公司)、硅胶(200~300目,山东烟台化学工业研究院)。
1.2 仪 器
单晶X射线衍射实验在Bruker SMART APEX CCD型单晶衍射仪上进行。核磁共振谱在DRX‑500型核磁共振仪上测试(氢谱:500 MHz,碳谱:125 MHz),以四甲基硅烷(TMS)为内标,氘代氯仿(CDCl3)或二甲基亚砜(DMSO‑d6)为溶剂。元素分析在VARIO EL型元素分析仪上测定。光致发光光谱在F‑7000型荧光分光光度计上测试。紫外可见光谱在Varian Carry 50型紫外可见分光光度计上测试。
1.3 配合物的合成与表征
(2,4‑2R‑mpq)2Ir(tmd)铱磷光配合物的合成路线如图1所示。

图1 配合物1~3的合成路线Fig.1 Synthetic routes for complexes 1‑3
1.3.1 (2,4‑2F‑mpq)2Ir(tmd)(1)的合成
称取四(2,4‑二氟苯基‑4‑甲基喹啉)二氯化铱二聚体(5.0 g,3.40 mmol)、tmd(3.13 g,17.00 mmol)和无水碳酸钠(3.60 g,34.00 mmol)置于500 mL三颈圆底烧瓶中,加入150 mL乙二醇单乙醚,反复除气3次。在氩气气氛下,加热回流反应4 h。冷却,过滤,在60℃下真空干燥。用二氯甲烷快速过柱,将二氯甲烷浓缩至200 mL时,往旋蒸瓶中加入50 mL无水乙醇,旋蒸除去二氯甲烷,过滤、干燥得到黄色固体4.55 g,收率为83.7%。1H NMR(500 MHz,CDCl3):δ 7.92(s,2H),7.90(d,J=1.2 Hz,2H),7.50~7.46(t,J=7.5 Hz,2H),(ddd,J=8.3,6.8,1.3 Hz,4H),6.46~6.41(ddd,J=12.8,9.3,2.3 Hz,2H),6.09~6.06(dd,J=8.8,2.3 Hz,2H),4.86(s,1H),2.89(s,6H),0.60 s,18H)。13C NMR(125 MHz,CDCl3):δ 194.25,166.86,166.80,163.26,163.16,162.72,162.62,161.23,161.13,160.66,160.56,155.36,155.30,148.23,146.77,131.31,130.36,127.19,126.45,125.92,123.44,120.71,120.56,118.19,118.17,118.06,118.04,97.49,97.27,97.06,89.21,40.59,27.69,19.34。元素分析按 C43H29N2F4O2Ir的计算值(%):C,59.10;H,3.34;N,3.21。实测值(%):C,59.12;H,3.36;N,3.24。
1.3.2 (2,4‑2MeO‑mpq)2Ir(tmd)(2)的合成
用四(2,4‑二(甲氧基)苯基‑4‑甲基喹啉)二氯化铱二聚体(4.90 g,3.40 mmol)替换四(2,4‑二氟苯基‑4‑甲基喹啉)二氯化铱二聚体(5.0 g,3.40 mmol),得到红色固体4.08 g,收率为74.6%。1H NMR(500 MHz,CDCl3):δ 8.72~8.71(d,J=0.7 Hz,4H),8.31~8.29(d,J=8.9 Hz,2H),7.81~7.79(dd,J=8.2,1.1 Hz,2H),7.39~7.33(m,2H),6.08~6.07(t,J=3.5 Hz,2H),5.85~5.84(d,J=2.3 Hz,2H),4.77(s,1H),3.99(s,6H),3.28(s,6H),2.82(s,6H),0.60(s,18H)。13C NMR(125 MHz,CDCl3):δ 193.55,169.13,160.23,159.79,157.64,148.44,144.34,129.22,128.99,127.60,125.72,124.35,122.82,121.45,112.87,92.25,88.49,55.08,54.65,40.48,27.79,19.39。元素分析按C47H51N2O4Ir的计算值(%):C,62.71;H,5.71;N,3.11。实测值(%):C,62.72;H,5.69;N,3.12。
1.3.3 (2,4‑2CF3‑mpq)2Ir(tmd)(3)的合成
用四(2,4‑二(三氟甲基)苯基‑4‑甲基喹啉)二氯化铱二聚体(6.37 g,3.40 mmol)替换四(2,4‑二氟苯基‑4‑甲基喹啉)二氯化铱二聚体(5.0 g,3.40 mmol),得到红色固体5.73 g,收率为77.7%。1H NMR(500 MHz,CDCl3):δ 8.38(s,2H),7.99~7.93(m,4H),7.63(s,2H),7.55~7.51(dd,J=33.6,26.2 Hz,2H),7.34~7.30(m,2H),6.97(s,2H),4.77(s,1H),2.90(s,6H),0.59(s,18H)。13C NMR(125 MHz,CDCl3):δ 194.46,166.23,155.86,148.33,148.11,146.61,136.67,130.61,127.90,127.65,127.45,127.22,127.12,126.68,125.26,124.33,123.55,123.09,122.16,121.85,121.78,121.72,121.65,117.86,88.99,40.60,27.73,19.56。 元 素 分 析 按C47H39N2F12O2Ir的计算值(%):C,52.08;H,3.63;N,2.58。实测值(%):C,52.05;H,3.65;N,2.60。
1.4 单晶X射线结构测试和解析
在室温下,将适量的3个铱(Ⅲ)配合物分别溶于二氯甲烷中,加入适量的无水乙醇,过滤除去不溶物,密封,随着二氯甲烷的缓慢挥发,分别制备得到配合物1~3的单晶。选取大小为0.960 mm×0.250 mm×0.180 mm(1)、0.13 mm×0.11 mm×0.1 mm(2)、0.320 mm×0.270 mm×0.230 mm(3)的配合物单晶,安装在Bruker APEX‑ⅡCCD型单晶X射线衍射仪上,使用经石墨单色器化的Mo Kα射线(λ=0.071 073 nm),以θ‑ω扫描方式在设定的2θ角度范围(1.195°~31.547°(1)、2.277°~26.022°(2)、1.666°~25.998°(3))内收集衍射数据。衍射数据用SADABS程序进行经验吸收校正,它们的晶体结构都是在Olex2上用SHELXTL‑2018程序解析和精修,对所有非氢原子坐标及各向异性温度因子进行全矩阵最小二乘法修正至收敛,氢为理论加氢。
CCDC:2217725,1;2217726,2;2217727,3。
2 结果与讨论
2.1 配合物的晶体结构
配合物1~3的晶体学数据、主要键长和键角分别列于表1和表2中,图2给出了3个配合物的分子结构。由表1可知3个配合物的晶体均属于三斜晶系,空间群为P1。由表2中的键长和键角数据,结合图2的分子结构,可以看出3种苯基喹啉类铱配合物分子均呈稍微扭曲的八面体构型,中心铱(Ⅲ)分别与2个主配体2,4‑2R‑mpq的C原子和N原子配位形成2个稳定五元螯合环,同时,还和tmd的2个O原子配位形成1个稳定的六元螯合环,主配体中的2个配位碳原子为顺式构象,2个配位氮原子则采取反式构象。
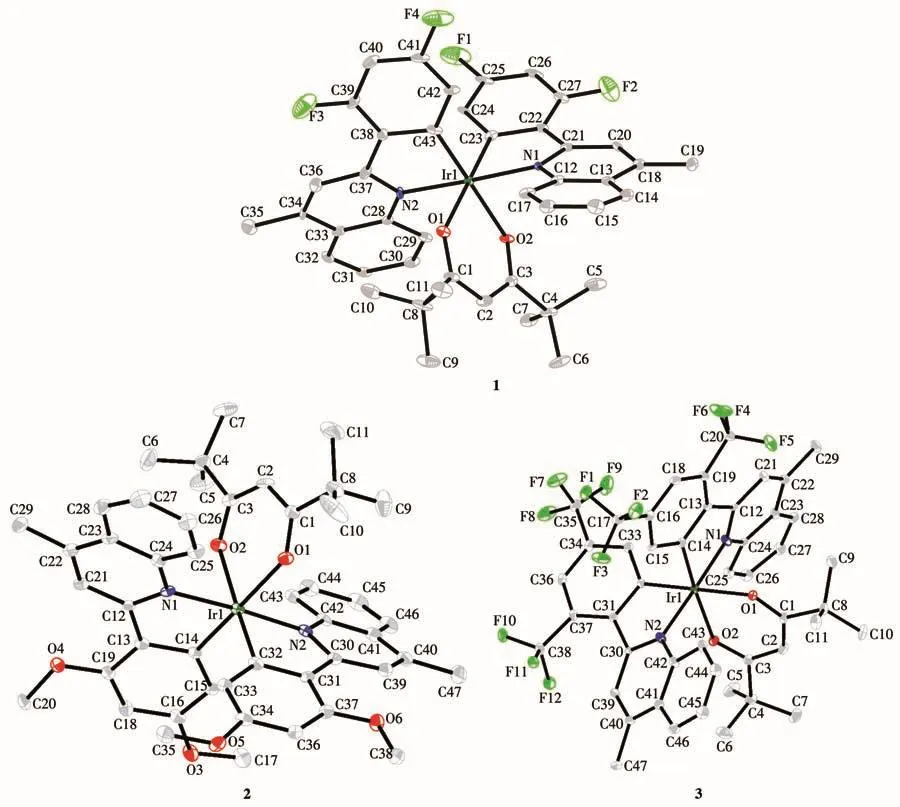
图2 配合物1~3的椭球率20%的晶体结构Fig.2 Crystal structures of complexes 1‑3 with an ellipsoid probability of 20%
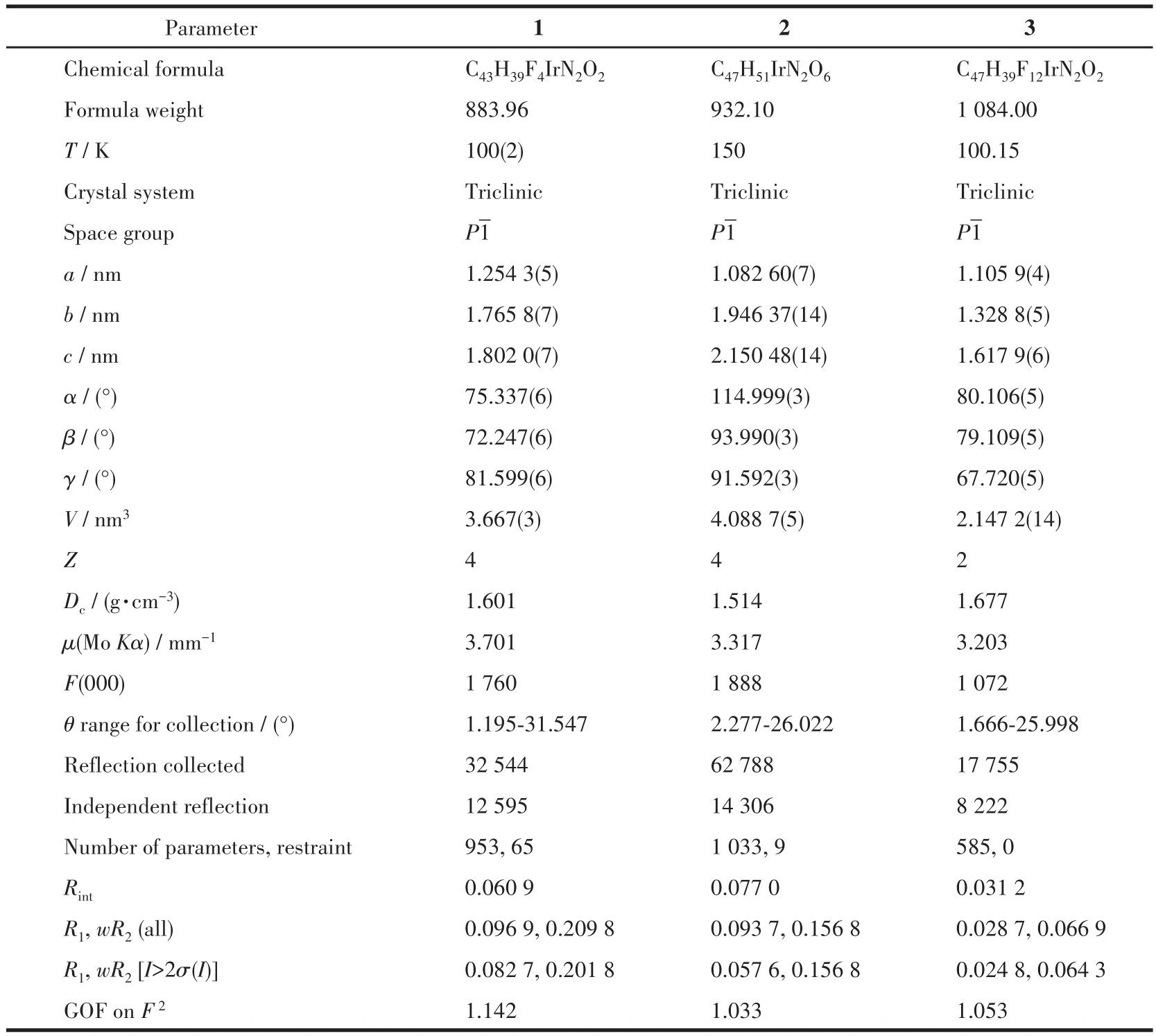
表1 配合物1~3的晶体学数据Table 1 Crystallographic data for complexes 1⁃3

表2 配合物1~3的主要键长(nm)和键角(°)Table 2 Selected bond lengths(nm)and angles(°)of complexes 1⁃3
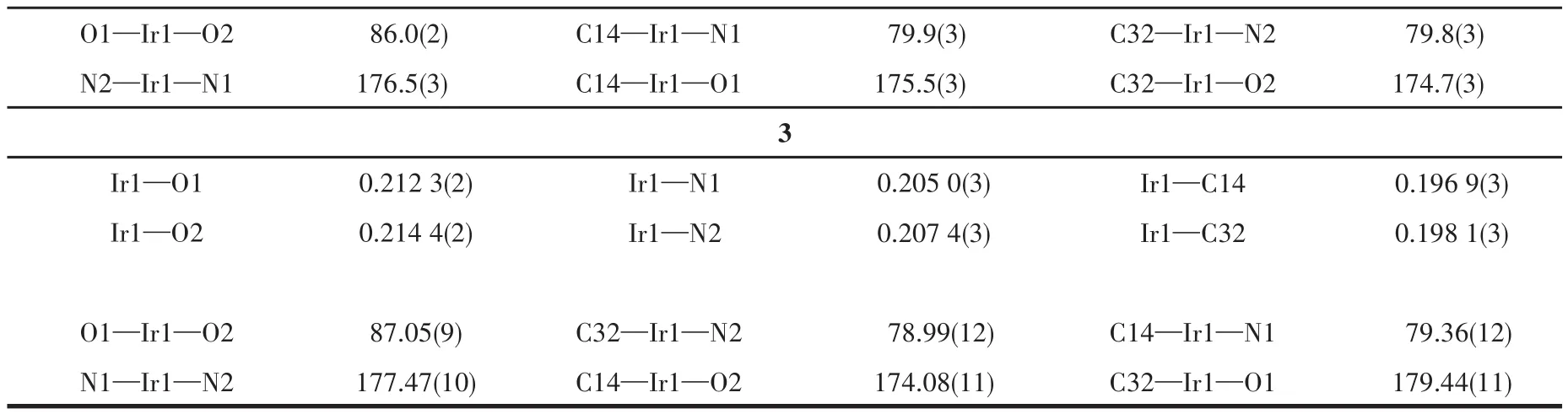
续表2
2.2 配合物的光物理性能
室温条件下,在10 µmol·L−1的二氯甲烷溶液中测试了配合物1~3的紫外可见光谱(图3)和光致发光光谱(图4),对应的主要光物理性能参数列于表3中。由图3可以看出,通过在苯基‑4‑甲基喹啉中苯基的2位和4位引入氟、甲氧基、三氟甲基,制备得到的3种铱磷光配合物的紫外可见吸收光谱比较相似,在260~290 nm范围内有强的特征吸收峰,可归属配体自旋允许1LC(1π‑π*)的跃迁。在300~500 nm范围内有较弱的吸收峰,该吸收源自金属的单重态和三重态到配体的电荷转移(1MLCT和3MLCT)跃迁。尽管3个铱磷光配合物具有相似的吸收特征,但吸收峰的位置上存在较大的差异,与甲氧基取代铱磷光配合物2相比,含氟取代基的铱磷光配合物1的吸收峰位置发生了蓝移,含氟数量多的三氟甲基取代后得到的铱磷光配合物3的吸收峰位置蓝移更明显。这主要是受氟原子吸电子诱导效应的影响[11‑12]。
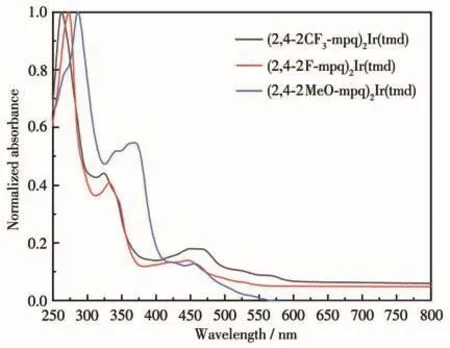
图3 (2,4‑2R‑mpq)2Ir(tmd)的紫外可见吸收光谱Fig.3 Absorption spectra of(2,4‑2R‑mpq)2Ir(tmd)
由图 4和表3可以看出,(2,4‑2F‑mpq)2Ir(tmd)(1)、(2,4‑2MeO‑mpq)2Ir(tmd)(2)和(2,4‑2CF3‑mpq)2Ir(tmd)(3)的发射波长分别为570、582和604 nm,除氧溶液中的量子产率分别为96%、80%和80%。结果表明,在苯基‑4‑甲基喹啉中苯基的2位和4位分别引入氟、甲氧基和三氟甲基后,由于氟和三氟甲基均为吸电子基团,配合物2的发射波长介于氟和三氟甲基取代苯基喹啉类铱磷光配合物1和3中间,也可以推断出甲氧基在这里体现出吸电子效应,为吸电子基团。这与传统认知中甲氧基体现为供电子基团不一致。前已述及,甲氧基表现出供电子还是吸电子效应,主要与甲氧基所处的位置有关。

表3 配合物1~3的光物理性能参数Table 3 Photophysical performance parameters of complexes 1⁃3
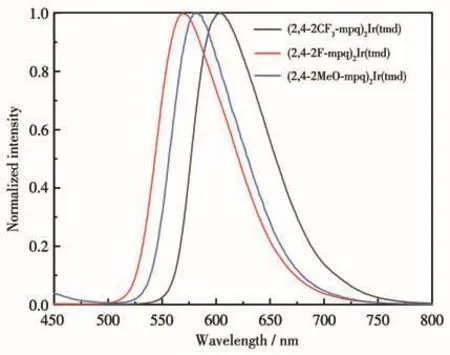
图4 (2,4‑2R‑mpq)2Ir(tmd)的光致发光光谱Fig.4 Photoluminescence spectra of(2,4‑2R‑mpq)2Ir(tmd)
2.3 配合物的理论计算分析
为了详细分析配合物的电子结构和光谱性质,我们进行了详细的理论计算。全部计算是利用Gaussian16程序中的密度泛函方法完成的。在根据报道和泛函选择分析基础上,从配合物的解析结构出发,最终采用B3LYP/LanL2TZ的方法优化了3个配合物的基态构型,并得到了它们的分子轨道分布特征(图5)。3个配合物具有相似的电子云分布特征,其HOMO主要定域于主配体的苯环和中心金属铱(Ⅲ)上,LUMO能级主要定域于主配体的喹啉环上。但取代基对电子云分布有一定的影响,CF3取代使得配合物3的电子云较分散,发光波长会向长波方向移动;F和MeO取代后,配合物的电子云聚集,发光向短波方向移动。三氟甲基取代后配合物的发射波长大于氟和甲氧基取代后配合物的发射波长,这与光致发光光谱获得的发射波长的变化趋势是一致的。
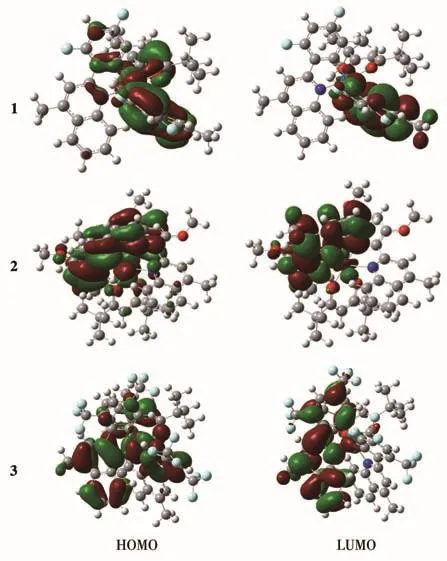
图5 铱磷光配合物1~3的HOMO‑LUMO能级图Fig.5 HOMO‑LUMO energy level diagram for iridium phosphorescent complexes 1‑3
3 结论
以2,2,6,6‑四甲基庚二酮(tmd)为辅助配体,2,4‑二取代基苯基‑4‑喹啉(2,4‑2R‑mpq)为主配体,在主配体苯基的2位和4位同时引入氟(F)、甲氧基(MeO)或三氟甲基(CF3),成功合成出3个吸电子基团修饰的苯基喹啉类铱磷光配合物(2,4‑2R‑mpq)2Ir(tmd)(R=F、MeO、CF3)。利用X射线单晶衍射仪测定了3个配合物的晶体结构,晶体均属于三斜晶系,空间群为P1。光物理性能研究结果表明:3个铱(Ⅲ)配合物具有相似的吸收特征,不同的吸电子基团导致了吸收峰位置不同,含氟取代基吸收峰位置发生蓝移,含氟数量越多,蓝移越明显。吸电子基团对配合物的电子云分布有一定影响,CF3取代使得配合物的电子云较分散,发光波长向长波方向移动,而F和MeO取代后,配合物的电子云聚集,发光向短波方向移动。(2,4‑2F‑mpq)2Ir(tmd)、(2,4‑2MeO‑mpq)2Ir(tmd)和(2,4‑2CF3‑mpq)2Ir(tmd)的发射波长分别为570、582和604 nm,除氧溶液中的量子产率分别为96%、80%和80%,有作为有机电致发光材料的应用潜力。
