不同产地独一味总黄酮的质量评价 Δ
2023-02-27陈瑞鑫蒋运斌陈文莉康点点李蕊蒋桂华成都中医药大学药学院西南特色中药资源国家重点实验室成都67西南大学药学院中医药学院重庆40075成都中医药大学国学院成都67
陈瑞鑫 ,蒋运斌 ,陈文莉 ,康点点 ,李蕊 ,蒋桂华 , (.成都中医药大学药学院/西南特色中药资源国家重点实验室,成都 67;.西南大学药学院中医药学院,重庆 40075;.成都中医药大学国学院,成都67)
独一味为唇形科植物独一味Lamiophlomis rotata(Benth.)Kudo的干燥地上部分,藏药名为“达巴”,系藏族习用药材,也常作为中药使用。2020年版《中国药典》载其具有活血止血、祛风止痛的功效,可用于治疗跌打损伤、风湿痹痛、黄水病等。独一味具有多种化学成分,主要包括环烯醚萜类、黄酮类、苯乙醇苷类以及多糖类[1]。黄酮类成分为广泛存在于自然界植物中的次生代谢产物,现代药理学研究表明该类成分具有抗炎、抗氧化、抗病毒、抗肿瘤、抗菌等多种药理活性[2]。目前已有学者发现独一味黄酮类成分具有较好的抗血小板聚集活性[3]和抗炎作用[4]。结合独一味功效,本课题组前期通过网络药理学研究发现独一味黄酮类成分可能在其抗类风湿性关节炎的过程中发挥了重要作用,特别是木犀草素[5];且现代药理学研究表明木犀草素具有抗类风湿性关节炎的活性[6]。为确证网络药理学的预测结果,本课题组前期基于佐剂性关节炎大鼠模型进行了相关研究,结果表明独一味黄酮类成分是独一味抗类风湿性关节炎的关键药效物质基础之一[7]。
独一味总黄酮具有潜在的药效,但目前国内外学者研究独一味主要围绕其环烯醚萜类成分进行[8―9],对其黄酮类成分的研究较为匮乏,不利于从中医药整体观角度对独一味质量进行评价和控制。为有效控制独一味黄酮类提取物的质量,本研究通过渗漉法提取、聚酰胺柱纯化了15批不同产地独一味的总黄酮,采用紫外分光光度法对其含量进行测定,通过高效液相色谱(HPLC)法对其指纹图谱进行研究,并运用化学计量学对不同产地独一味总黄酮的质量进行评价,旨在为独一味总黄酮质量评价奠定基础。
1 材料
1.1 主要仪器
本研究所用主要仪器有BP121S型电子分析天平[赛多利斯科学仪器(北京)有限公司]、DZKW-4型电子恒温水浴锅[中兴伟业仪器(北京)有限公司]、RE-5203型旋转蒸发器[亚荣生化仪器(上海)厂]、KQ-500VDE型双频数控超声波清洗器[超声仪器(昆山)有限公司]、UEF1010033型紫外-可见分光光度计[美谱达仪器(上海)有限公司]、Ultimate3000型HPLC仪[赛默飞世尔科技(美国)公司]。
1.2 主要药品与试剂
木犀草苷对照品(批号MUST-21111817,纯度≥99.0%)、木犀草素对照品(批号MUST-21072311,纯度≥98.0%)均购于成都曼思特生物科技有限公司;芦丁对照品(批号AF21020854,纯度≥98.0%)购于成都埃法生物科技有限公司;甲醇、乙腈均为色谱纯;亚硝酸钠、硝酸铝、氢氧化钠及乙醇均为分析纯;水为超纯水。15批独一味药材经成都中医药大学药学院蒋桂华教授鉴定均为唇形科植物L. rotata(Benth.)Kudo的干燥地上部分,样品来源信息见表1。
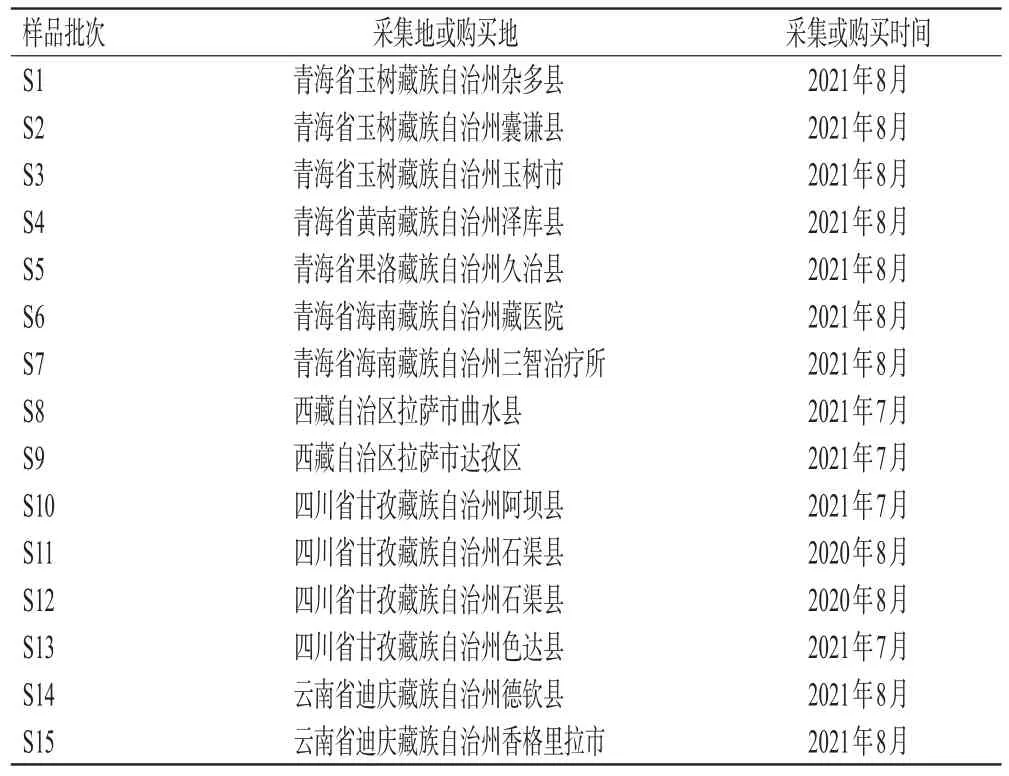
表1 独一味样品来源信息
2 方法与结果
2.1 独一味总黄酮的制备
根据相关文献[10]及前期工作基础,取独一味粉末100 g加入渗漉桶中,加入95%乙醇没过粉末,静置24 h后开始提取。95%乙醇适当流速渗漉,收集渗漉液,于50 °C减压旋转蒸发回收溶剂,待回收约30 mL时将浓缩的渗漉提取液与预处理的聚酰胺粉(50 g)混匀,50 °C水浴蒸干,上样于已处理好的聚酰胺层析柱中,放置24 h。先以高纯水(3倍柱体积)洗脱至无色,然后用70%乙醇溶液(14倍柱体积)洗脱,并收集70%乙醇洗脱液,减压浓缩,即得独一味总黄酮。
2.2 独一味总黄酮的含量测定及纯度计算
采用紫外分光光度法进行独一味总黄酮的含量测定,并计算其纯度,含量测定方法参考2020年版《中国药典》独一味片总黄酮含量测定项方法。
2.2.1 供试品溶液的制备 取制备的独一味总黄酮样品50 mg,精密称定,置于50 mL容量瓶中,加70%乙醇30 mL,超声(功率200 W,频率60 kHz,下同)使溶解,放冷,加70%乙醇至刻度,摇匀,即得。
2.2.2 对照品溶液的制备 取芦丁对照品10 mg,精密称定,置于50 mL容量瓶中,加70%乙醇30 mL,超声使溶解,放冷,加70%乙醇至刻度,摇匀,即得。
2.2.3 线性关系的考察 准确吸取对照品溶液1.0、2.0、3.0、4.0、5.0、6.0 mL,分别放入25 mL容量瓶中,用70%乙醇溶液补充至10 mL,然后加入1 mL 5%亚硝酸钠溶液,摇匀后静置6 min,加1 mL 10%硝酸铝溶液,摇匀,静置6 min,再加10 mL 1 mol/L氢氧化钠溶液,用70%乙醇定容至25 mL,反应15 min后,以相应的溶液为空白参照,在其最大吸收波长处测定系列浓度下的吸光度。以芦丁质量浓度(μg/mL)为横坐标(X),吸光度为纵坐标(Y)绘制标准曲线,得到回归方程为Y=10.674X,R2=0.998 8,线性范围为8.0~48.0 μg/mL。结果表明,当芦丁质量浓度在8.0~48.0 μg/mL范围内线性关系良好。

2.3 独一味总黄酮HPLC指纹图谱研究
2.3.1 色谱条件 以 Waters Symmetry C18(250 mm×4.6 mm, 5 μm)为色谱柱;以乙腈(A)-0.2%甲酸溶液(B)为流动相进行梯度洗脱(0~15 min,8%A→13%A;15~30 min,13%A;30~40 min,13%A→17%A;40~75 min,17%A→38%A;75~76 min,38%A→8%A;76~85 min,8%A);流速为1.0 mL/min;柱温为35 ℃;检测波长为245 nm;进样量为10 μL。记录75 min的色谱图。
2.3.2 对照品溶液的制备 分别精密称取木犀草苷、木犀草素对照品各适量,置于10 mL容量瓶中,加甲醇定容,混匀,配制成0.3 mg/mL的对照品溶液,过0.22 μm微孔滤膜,即得。
2.3.3 供试品溶液的制备 精密称取独一味总黄酮样品0.1 g,置于25 mL容量瓶中,加甲醇超声使溶解,定容,混匀,过滤,取续滤液过0.22 μm微孔滤膜,即得。
2.3.4 精密度考察 精密称取样品(S1)0.1 g,按“2.3.3”项下方法制备供试品溶液,按“2.3.1”项下色谱条件连续进样测定6次。结果显示,以木犀草苷为参照峰,各共有峰相对保留时间RSD在0.06%~0.19%之间,相对峰面积RSD在0.14%~3.84%之间,表明仪器精密度良好。
2.3.5 稳定性考察 按照“2.3.3”项下方法制备供试品溶液(S1),分别在制备后在室温放置0、4、8、12、18、24 h时按“2.3.1”项下色谱条件进样测定。结果显示,以木犀草苷为参照峰,各共有峰相对保留时间RSD在0.07%~0.49%之间,共有峰相对峰面积RSD在0.05%~5.50%之间,表明供试品溶液在室温放置24 h内稳定性良好。2.3.6 重复性考察 按照“2.3.3”项下方法平行制备6份供试品溶液(S1),按“2.3.1”项下色谱条件进样测定。结果显示,以木犀草苷为参照峰,各共有峰相对保留时间RSD在0.04%~0.22%之间,相对峰面积RSD在0.55%~3.52%之间,表明方法重复性良好。
2.3.7 指纹图谱的建立及样品分析 按照“2.3.3”项下方法制备15批独一味总黄酮供试品溶液,并按“2.3.1”项下色谱条件进样测定,记录色谱图,导入《中药色谱指纹图谱相似度评价系统(2012版)》软件。设置S4样品图谱为参照图谱,选择中位数法,时间窗口宽度为0.1 min,通过多点校正和Mark峰匹配生成指纹图谱的叠加图谱及对照指纹图谱(R),结果见图1;15批独一味总黄酮中共确定了5个共有峰,其中3号峰为木犀草苷,详见图2。

图1 15批独一味总黄酮HPLC叠加图谱及对照指纹图谱(R)
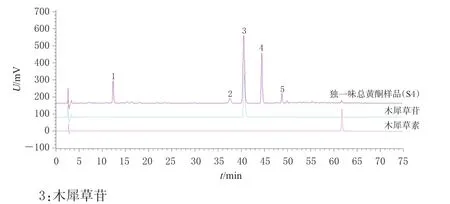
图2 独一味总黄酮样品和木犀草苷、木犀草素对照品的HPLC图
由图1显示,3号峰(木犀草苷)是独一味总黄酮中主要含有的成分,其苷元即为木犀草素。部分独一味总黄酮色谱图中虽出现了木犀草素的峰,但其峰面积较小。有些批次如S2、S9、S10独一味总黄酮的色谱图中未出现木犀草素的峰,这可能是由于天然黄酮类化合物多以苷类形式存在[11],且有研究表明,黄酮苷类化合物口服后常被肠道菌群产生的水解酶代谢以苷元的形式吸收入血[12]。木犀草苷作为共有峰能为总黄酮质量控制提供较大贡献。
2.3.8 相似度评价 将15批独一味总黄酮色谱图导入《中药色谱指纹图谱相似度评价系统(2012版)》软件,计算其与对照指纹图谱的相似度。结果显示,S1~S15批次独一味总黄酮与对照指纹图谱的相似度分别为0.999、0.953、0.999、0.999、0.997、1.000、1.000、0.983、0.925、0.992、0.995、0.991、0.996、0.996、0.976,表明15批独一味总黄酮的质量稳定,但仍然存在差异。
2.4 独一味总黄酮的化学模式识别分析
2.4.1 聚类分析 以5个共有峰峰面积为变量,利用SPSS 20.0软件,先对原始数据进行标准化,再采用组间平均数联结法进行聚类分析,结果见图3。当判别距离为15时,15批独一味总黄酮可聚为4类,S1、S3~S9为第1类,为青海省和西藏自治区样品;S14、S15为第2类,均为云南省样品;S10~S13为第3类,均为四川省样品;S2为第4类,表明15批独一味总黄酮的质量存在一定的差异性,具有明显地域性。

图3 15批独一味总黄酮的聚类分析图
2.4.2 主成分分析 将15批独一味总黄酮色谱图中5个共有峰的相对峰面积导入SPSS 20.0软件进行主成分分析,分析得到的特征值及方差贡献率见表2。根据特征值>1,共提取到2个主成分,包含了样品指纹图谱中74.031%的信息。

表2 15批独一味总黄酮的特征值及累计方差贡献率
为更直观地评价各产地独一味总黄酮的质量,采用方差贡献率对15批独一味总黄酮进行综合评价,主成分1和主成分2的得分计算公式分别为:y1=0.549x1+0.476x2+0.399x3+0.534x4+0.196x5,y2=-0.255x1+0.920x2-0.462x3+0.233x4+0.811x5,其中 y1和 y2代表主成分1、2 的得分,x1~x5为5个共有峰的峰面积,y1和y2的各项系数为主成分1、2的载荷值与特征值开平方的比值;按照公式“y=0.513 54y1+0.226 77y2”计算综合得分(y),y值越高表示独一味总黄酮的质量越好。结果见表3。由综合得分排序和聚类分析可知,第1类和第4类即青海省和西藏自治区的独一味总黄酮质量较优。

表3 15批独一味总黄酮主成分分析综合得分表
2.4.3 正交偏最小二乘法-判别分析 将15批独一味总黄酮5个共有峰峰面积导入SIMCA-P 14.1软件,通过正交偏最小二乘法-判别分析(orthogonal partial least squares-discriminant analysis,OPLS-DA),根据其变量重要性投影(variable importance in the projection,VIP)参数进行分析,结果见图4。以VIP值>1作为标准[13],由图可知,2、5、3号峰(木犀草苷)VIP值均大于1,表明这3个成分为导致15批独一味总黄酮差异性的主要成分。

图4 15批独一味总黄酮OPLS-DA的VIP图
3 讨论
本课题组前期对相关文献[11,14]记载的独一味总黄酮提取方法进行了比较,发现聚酰胺树脂对独一味总黄酮的吸附、解吸效果优于D-101大孔树脂。在此基础上本研究结合相关文献考察的洗脱条件提取了15批不同产地独一味的总黄酮,通过紫外分光光度法测定其含量,计算得到15批独一味总黄酮纯度平均值为77.72%,表明15批独一味总黄酮纯度较高,可用于独一味总黄酮指纹图谱研究。15批独一味总黄酮含量差异较大,可能是聚酰胺树脂对不同类型黄酮化合物的吸附作用不同以及各批次独一味样品中不同类型黄酮化合物含量有差异造成的[15]。
本研究前期考察了甲醇-水、乙腈-水、乙腈-0.2%甲酸溶液和甲醇-0.2%甲酸溶液4种流动相以及不同流动相梯度对独一味总黄酮指纹图谱的影响,以基线平稳、色谱峰多且分离好为标准,最终选择了乙腈-0.2%甲酸溶液作为流动相。
由指纹图谱共有峰相关分析和相似度评价可知,15批独一味总黄酮的相对峰面积RSD较大,相似度虽然在0.925~1.000之间,但各批次间仍然存在差异。为进一步明确引起差异性的主要原因,本研究采用聚类分析、主成分分析和OPLS-DA 3种化学计量学方法对15批独一味总黄酮进行质量评价。聚类分析表明,15批独一味总黄酮可根据产地聚为4类,表明地域性可引起独一味总黄酮的质量差异;通过主成分分析进一步得到第1类和第4类产地即青海省和西藏自治区的独一味总黄酮质量较好,与《中国植物志》[16]记载的独一味主产区一致;通过OPLS-DA筛选出3个影响独一味总黄酮质量的主要成分,其中1个成分为木犀草苷,也暗示木犀草苷等3个主要成分的含量高低与独一味总黄酮的质量密切相关。
综上所述,青海省和西藏自治区的独一味总黄酮质量较好。本研究可为不同产地独一味总黄酮的质量评价提供参考依据。
