强直性脊柱炎发病机制及药物调控研究进展
2022-12-16张曙琼邵粉丽
张曙琼,邵粉丽,孙 洋
(南京大学生命科学学院生物技术与药学系,医药生物技术国家重点实验室,江苏 南京 210023)
强直性脊柱炎(ankylosing spondylitis,AS)是一种慢性炎症性风湿病,具有遗传特性、男性多发和年轻化等特征。AS早期以骶髂关节和脊柱等中轴关节炎症引起的炎性背痛为主要表现,疾病末期出现脊柱关节融合现象。同时,超半数的AS患者外周关节受累。此外,AS患者常伴随关节外疾病,如急性葡萄膜炎和骨质疏松症等[1-2]。
修订后的纽约标准将骶髂关节放射性进展合并包括炎性背痛、腰椎活动受限和胸部扩张受限在内的任一临床特征作为AS诊断标准。非甾体抗炎药反应性,人类白细胞抗原-B27(human leukocyte antigen-B27,HLA-B27)阳性以及影像学表现可能也是AS发生的指征。传统的影像学检测常采用X光照射,但其只能检测炎症的结构性结果,而早期炎症无慢性结构变化,因此可借助核磁共振来检测早期关节中的炎症[2]。此外,计算机断层扫描影像也可以检测到AS患者脊柱的经典“竹节”样病变[3]。
本文将对近年来AS发病机制的研究及药物调控进展进行概述。
1 发病机制
1.1 遗传因素遗传因素被认为是AS的致病因素。对AS患者中人类白细胞抗原(human leukocyte antigen,HLA)及其它主要组织相容性复合体(major histocompatibility complex,MHC)分子和非MHC分子的大量研究也表明了遗传因素与AS的强相关性[4]。遗传因素的影响也导致了AS的家族聚集性。
1.1.1HLA-B27及其它HLAⅠ类分子 目前最受认可的与AS相关的基因是HLA-B27,90%~95%的AS患者呈HLA-B27阳性[5]。Meta分析显示HLA-B27基因多态性B2702、B2704、B2705、B2715可能是AS的风险因素,而B2703、B2706、B2707、B2727、B2729和B2747可能是AS的保护因素[6]。
从机制上,关节炎多肽由HLA-B27向CD8+T淋巴细胞呈递,触发CD8+T淋巴细胞。内质网氨基肽酶1/2(endoplasmic reticulum aminopeptidase1/2,ERAP1/2)的错误剪切会产生异常的多肽,导致HLA-B27游离重链和同源二聚体在细胞膜上循环,抗原呈递细胞将HLA-B27二聚体呈递给杀伤细胞免疫球蛋白样受体(killer-cell immunoglobulin-like receptors,KIR)和白细胞免疫球蛋白样受体,AS患者KIR3DL2+CD4+T淋巴细胞增殖,激活NK细胞和Th17细胞,IL-17产生增加,这些细胞主要分泌肿瘤坏死因子-α(tumor necrosis factor-α,TNF-α)和干扰素-γ(interferon-γ,IFN-γ)。IL-17与TNF-α或IFN-γ协同作用,刺激炎症因子的释放,影响骨结构,从而在AS的发生发展中发挥作用[7-9]。
除HLA-B27外,HLA-B07、HLA-B57、HLA-B51、HLA-B47、HLA-B40、HLA-B13等HLA-B等位基因与AS易感性也显著相关,且其它相关HLA分子可能使AS与其他疾病相关联。例如,HLA-B51同为AS和风湿免疫病贝赫切特病(Behcet disease)的主要风险基因。HLADRB1*0103同为AS和克罗恩病(Crohn’s disease)的主要风险基因之一,与克罗恩病患者常并发AS相吻合[4]。此外,HLA-B60会增加AS的风险,HLA-B39也与HLA-B27阴性AS具有相关性[5]。
1.1.2ERAP1/2 内质网氨基肽酶(endoplasmic reticulum aminopeptidase,ERAP)与AS发生密切相关。ERAP1与AS相关的HLA Ⅰ类分子HLA-B27和HLA-B4001相互作用,所以与HLA-B27/HLA-B4001阳性的AS有关,而ERAP2与HLA-B27阴性/阳性的AS均有关[10]。ERAP1/2参与了内质网中的肽剪切,改变了HLA I类分子呈递的肽的长度和氨基酸组成。ERAP1/2异常剪切产生的异常多肽-HLA-B27复合物聚集在内质网,触发未折叠蛋白反应、内质网应激、内质网相关降解和自噬[7]。
1.1.3其它AS相关遗传因素 除了最常见的HLA-B等位基因外,还有许多其它遗传因素与AS发病密切相关。真核翻译延长因子1ε1可能通过影响氨基酰tRNA的生物合成而参与AS的发生,而丝氨酸蛋白酶抑制剂家族A成员1可能通过参与血小板脱颗粒而参与AS的发生[6]。此外,以往的遗传学研究也证实,各种氨基肽酶基因与AS之间存在关联,如嘌呤霉素敏感型氨基肽酶基因、亮氨酸/半胱氨酰氨肽酶基因(leucyl/cystinyl aminopeptidase,LNPEP)。LNPEP和ERAP2同为内质网氨基肽酶家族的成员,在抗原交叉呈递中发挥剪切多肽的作用[5,11]。
1.2 免疫反应脊柱关节的慢性炎症会导致严重的慢性疼痛和僵硬,易导致脊柱强直和新骨形成。免疫系统的失调或过度激活对AS发病有重要影响,各种免疫细胞、分泌介质和标志物在AS的发病机制中发挥着重要作用[12]。
1.2.1炎症性肠病——肠道微生物 某些肠道病原体可以引发HLA-B27相关反应性关节炎,且炎症性肠病患者中AS和外周关节炎患病率大幅增加[1]。据报道,含有肠道菌群的HLA-B27转基因鼠会发生结肠炎及关节炎症,而在无菌环境中培育的转基因鼠则不会发生,再次证明了肠道微生物与炎症之间的关系[13]。
肠道微生物失调和先天免疫之间存在复杂的相互作用,这导致能够从肠道迁移到肠外部位的细胞的异常激活和扩增[11]。AS患者肠道中的炎症环境可损伤肠道上皮细胞,细菌入侵触发上皮细胞产生IL-33,IL-33调节巨噬细胞向M2表型的极化,而Paneth细胞与浸润的巨噬细胞产生IL-23[12]。此外,肠道微生物刺激树突状细胞和巨噬细胞产生IL-23,IL-23使Th17细胞和包括γδT细胞、恒定自然杀伤T细胞、ROR-γt+CD3+CD4-/CD8-T细胞、肥大细胞和中性粒细胞在内的先天免疫细胞产生IL-17、TNF、IL-22等炎性细胞因子,从而促进AS发生发展[14]。类似地,肠道微生物可以刺激单核细胞产生趋化Th17的促炎细胞因子IL-1β、IL-23、TNF等,在趋化因子的驱使下,分泌IL-1β的单核细胞进入关节,激活γδT细胞和恒定自然杀伤T细胞[15]。另外,AS动物模型中肠道稳态失调会导致炎性小体的激活,从而以IL-1β依赖性方式促使Ⅲ型细胞因子的产生[16]。
1.2.2炎性细胞因子——IL-23/Th17轴 IL-23/Th17轴在AS发病机制中占据举足轻重的地位。Th17和Th22细胞与几种应激的相互作用导致促炎分子IL-17、IL-22、TNF-α和IL-23的产生,而IL-23促进Th17细胞的分化和维持以及IL-17的产生,进而促进AS发病[7,14]。AS中IL-17A的下游致病作用可能与其影响中性粒细胞、巨噬细胞和上皮细胞的募集,以及随后释放其他促炎细胞因子如IL-1β、IL-6和TNF-α有关。IL-17F、IL-22、粒细胞-巨噬细胞集落刺激因子(granulocyte-macrophage colony stimulating factor,GM-CSF)和IL-9等其他17型相关细胞因子也可能参与AS炎症的发生[17]。
IL-17不仅由Th17细胞产生。在滑液中,巨噬细胞与髓过氧化物酶阳性细胞协同作用产生IL-23,含有KIR3DL2的NK细胞、γδT细胞、Th17和异常Ⅲ型先天淋巴细胞(type 3 innate lymphoid cells,ILC3)可响应IL-23产生IL-17。血液中肥大细胞、黏膜相关恒定T细胞和活化的恒定自然杀伤T细胞与炎性树突状细胞相互作用产生IL-17。ILC3存在于所有受累部位,并通过IL-23R对IL-23产生应答[12]。遗传学研究显示,参与IL-23/IL-17A通路的基因,如IL12B、RUNX3、EOME、TBX21、TYK2、CARD9、IL1R1、IL1R2、IL6R、IL7R、IL12B、IL27、NKX2和PTGER4等也与AS易感性相关[11]。
此外,炎性细胞因子在激活经典成骨通路中的积极作用也得到了证明,如IL-23/IL-17 增强Wnt、Hedgehog通路的活化[18],以及低强度TNF刺激通过核因子κB(nuclear factor kappa-B,NF-κB)和c-Jun氨基末端蛋白激酶/活化蛋白-1(c-Jun N-terminal kinase/ activator protein-1,JNK/AP-1)信号通路诱导Wnt蛋白的持续表达和随后的骨形成[19]。
IL-23/Th17轴在AS中的致病机制与其在银屑病中高度吻合[20],这也解释了AS为何常伴随银屑病发生。
1.2.3自身免疫靶点——Ⅱ型胶原、蛋白聚糖 虽然AS被认为是一种自身免疫病,但目前尚未有明确的证据表明其确切的自身免疫靶点。据报道,软骨蛋白聚糖和聚集蛋白聚糖可以诱导BALB/c小鼠发生脊柱炎和侵蚀性多发性关节炎,且针对蛋白聚糖的细胞免疫常见于AS患者[21]。此外,在AS患者的外周血和滑液样本中,HLA-B27限制性细胞毒性T淋巴细胞可以对蛋白聚糖和胶原衍生肽产生反应[1]。
1.3 成骨通路除了炎性背痛和关节炎破坏外,病理性新骨形成是AS的主要特征。因此,成骨通路对AS发病的影响不容忽视。
1.3.1Wnt/β-catenin通路 Wnt蛋白在骨稳态,特别是新骨形成过程中起着至关重要的作用,也在AS的病理性成骨中发挥作用[22]。有研究表明,AS中的高活动性炎症使骨诱导性Wnt蛋白促进异位新骨形成,此过程需要激活经典的Wnt/β-catenin和非经典的Wnt/PKCδ通路[19]。当Wnt蛋白与卷曲受体(frizzled,FZD)和低密度脂蛋白受体相关蛋白5/6(low density lipoprotein receptor related protein5/6,LRP5/6)共受体结合时,就会产生经典Wnt信号,从而阻止蛋白质与结肠腺瘤息肉易感蛋白(adenomatous polyposis coli,APC)、糖原合成酶激酶3β(glycogen synthase kinase-3β,GSK3β)和轴蛋白(Axin)的复合体对β-catenin进行标记降解,β-catenin转移到细胞核,与转录因子结合并激活目标基因的转录,促进成骨细胞分化和新骨形成[23]。此外,骨膜蛋白表达在AS患者中下调,并通过与Wnt信号相互作用促进成骨,这可能是AS发病机制之一[24]。
1.3.2BMP/Smads通路 骨形态发生蛋白(bone morphogenetic protein,BMPs)属于转化生长因子(transforming growth factor,TGF)超家族的成员,可以诱导异位软骨和骨形成,促进脊柱融合[25]。
BMPs与同源Ⅱ型受体结合,通过转磷酸化同源I型受体诱导Smad依赖和非Smad依赖的信号传导。在Smad依赖的信号通路中,磷酸化的R-Smad(pSmad1/5/8)复合物与Smad4共转位到细胞核中,招募辅助因子来调节成骨基因的表达,如Runx2、Dlx5和Osx[25]。此外,BMP还可以激活非Smad依赖的Wnt/β-catenin通路,共同影响骨形成[26]。
我们课题组意外发现,CD4-Cre介导的SHP2条件性敲除小鼠在7月龄以上会出现脊柱、髋关节和骶髂关节等中轴关节变形与僵直症状。通过T细胞转输、骨髓移植及使用Lck-Cre;SHP2f/f小鼠等实验,在排除T细胞和其他免疫细胞之后,我们使用mTmG荧光示踪技术证实CD4-Cre;SHP2f/f小鼠骨病变的诱因竟是骨骺生长板中软骨细胞上SHP2的缺失,导致小鼠软骨细胞稳态失调,表现为SOX9异常活化,骨骺生长板以及起止点等位置软骨细胞异常增殖并大量分泌BMP6,继而通过Smad1/5-Runx2通路促进成骨细胞分化,加剧异位骨化形成。这一机制在临床样本中也得到验证。该研究首次揭示了骨骺生长板闭合延迟可致AS骨病变的崭新发病机制,打破了AS发病机制的固有认知,使用老药索尼德吉恢复软骨细胞稳态可显著阻止AS的骨融合进程[27]。与此一致,早期AS患者韧带内可见软骨细胞分化,软骨形成后伴有钙化,钙化软骨引发的破骨细胞吸收产生了具有高水平活化TGF-β的骨髓,同时,BMP-pSmad1/5/8、BMP-pSmad2/3信号通路被激活,从而导致AS中病理性异位骨化[28]。
1.3.3Hedgehog通路 Hedgehog是软骨内骨形成过程中的关键调节因子,参与骨髓间充质干细胞(bone marrow mesenchymal stem cells,BM-MSCs)向成骨细胞的分化[29]。Indian hedgehog(IHH)是Hedgehog基因编码的同源蛋白之一,主要参与软骨细胞增殖和成熟,导致软骨内骨化,参与AS中新骨形成。临床数据表明,HLA-B27阳性个体的血清IHH水平明显高于HLA-B27阴性个体[30],体现了Hedgehog与AS间的相关性。此外,Hedgehog信号也作用于Wnt和BMP通路,共同促进软骨内成骨[31]。相似地,BM-MSCs中Hedge-hog信号的激活通过Wnt/β-catenin通路诱导软骨和骨肿瘤的形成[32]。
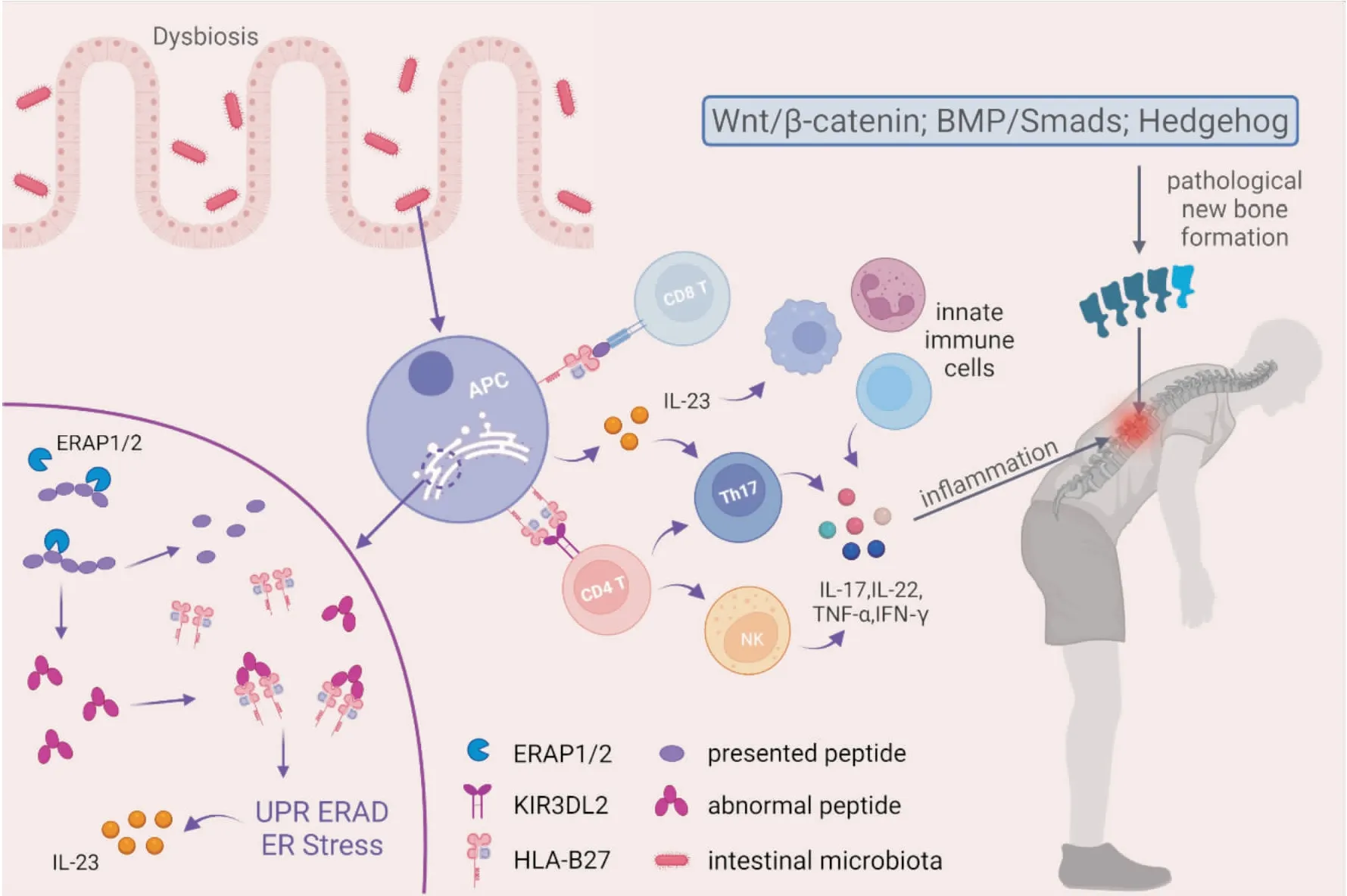
Fig 1 Main pathogenesis of AS
2 AS发病机制间联系
综上所述,AS的多种发病机制围绕HLA-B27展开,并相互联系,如Fig 1所示。自身反应性CD8+T细胞可能识别HLA-B27在抗原呈递细胞表面呈递的致关节炎肽;内质网中错误折叠的HLA-B27同源二聚体和异常剪切产生的多肽结合累积导致内质网应激,未折叠蛋白反应(unfolded protein response,UPR),内质网相关降解(endoplasmic reticulum-associated degradation,ERAD)等,进而导致IL-23的过度产生;细胞表面HLA-B27的异常表达导致与CD4+T细胞上的KIR3DL2等先天免疫受体相互作用,促进17型免疫应答;HLA-B27导致肠道菌群失调,从而影响抗原呈递细胞及其它先天免疫细胞,导致IL-17A、IL-22、TNF-α、IFN-γ和其他细胞因子的产生,从而促进AS的炎性环境[2,17]。同时,由经典成骨通路介导的病理性新骨形成也是AS重要的发病机制[27-28],这可能受炎性细胞因子的影响[18-19]。
3 药物治疗进展
目前临床上AS的治疗药物主要是通过控制症状和炎症、防止渐进性结构损伤、维护正常功能,最大限度地提高患者长期生活质量并使其正常参与社会活动。AS工作小组和欧洲风湿病联盟工作组的联合评估建议AS患者的最佳治疗需要结合非药物和药物治疗方式。药物治疗包括使用非甾体抗炎药、改善疾病的抗风湿病药物、镇痛药、局部和全身类固醇药物以及并发症治疗药物。非药物治疗包括教育、运动和理疗以及外科干预对AS进行治疗[33]。Tab 1展示了目前临床试验中正在研究的部分AS治疗药物。
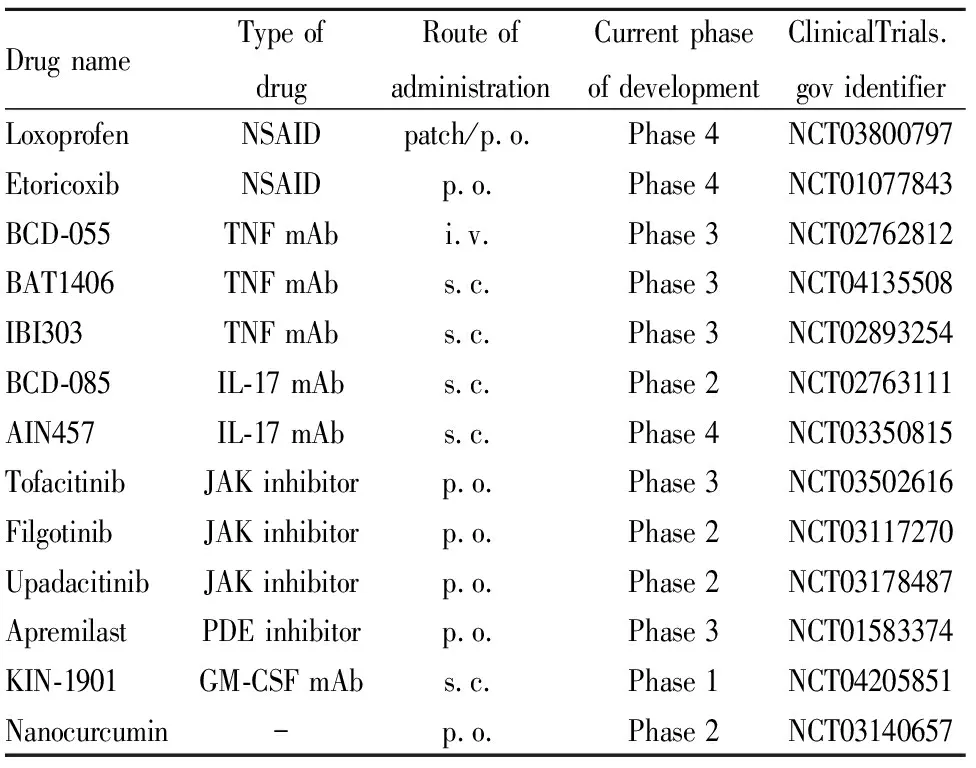
Tab 1 Main emerging therapies being investigated in AS clinical trials
3.1 非甾体抗炎药非甾体抗炎药(non-steroidal anti-inflammatory drugs,NSAIDs)是治疗AS常用的药物,其能有效减轻患者临床症状和体征,消除局部关节炎症,但不能控制疾病的活动和进展。常用于治疗AS的NSAIDs包括环氧化酶抑制剂双氯芬酸、吲哚美辛、酮洛芬、舒林酸、萘丁美酮和萘普生等[34]。对AS患者进行NSAIDs连续治疗可以在不显著增加毒性的情况下减缓放射学进展[35]。但非甾体抗炎药有胃肠道和心血管毒性作用,应限制使用[7]。
3.2 TNF抑制剂目前被FDA批准用于AS治疗的抗TNF药物主要包括英夫利昔单抗(Infliximab)、依那西普(Etanercept)、阿达木单抗(Adalimumab)、赛妥珠单抗(Certolizumab)和戈利木单抗(Golimumab)。这些药物可以改善疾病活动性和功能,并在短期内部分缓解AS临床症状[36]。此外,抗TNF-α治疗可以降低AS患者的血清IHH水平,并影响功能性Hedgehog通路靶基因的表达[37]。但长期使用TNF抑制剂对AS的疗效欠佳,且不能抑制AS的放射学进展[35],需要联用或更换其它药物。
3.3 IL-17、IL-23单克隆抗体最常用的IL-17A单抗苏金单抗(Secukinumab)已被证明在AS治疗中具有安全性和有效性,适用于对TNF阻滞剂治疗无效的患者。艾克司单抗(Ixekizumab)治疗也可有效改善AS患者放射学进展。另一种IL-17抑制剂布罗达单抗(Brodalumab)可以靶向IL-17受体,拮抗IL-17A同源二聚体、IL-17A/F异源二聚体等,其针对AS治疗的Ⅲ期临床试验已经完成[38]。比美吉珠单抗(Bimekizumab)是一种靶向IL-17A和IL-17F的双重抑制剂,可有效治疗AS[39]。而针对IL-23的两种抑制剂乌斯泰金单抗(Ustekinumab)和SKYRIZI(Risankizumab)的临床试验均未显示出对AS的疗效[7],这可能表明IL-23下游信号通路与AS的发病机制更相关。但与此结果相反,有研究表明Ustekinumab可有效缓解AS患者的症状[40]。
3.4 改善外周炎症药物AS是一种类风湿性疾病,可以使用抗风湿药物进行治疗。例如,柳氮磺胺吡啶、甲氨蝶呤、来氟米特对关节炎有一定的改善作用,但对放射性骨病变没有显著疗效。此外,关节内或关节周围注射糖皮质激素可以缓解AS中骶髂关节的疼痛[7,33]。
3.5 成骨通路抑制剂Wnt蛋白抑制剂Dickkopf-1(DDK-1)和硬化蛋白(sclerostin,SOST)结合LRP5/6共受体,抑制Wnt信号,导致胞浆中β-catenin的降解,从而抑制成骨细胞分化和骨增殖,而卷曲相关蛋白家族成员Cerberus和WIF作为拮抗剂可以直接与细胞外Wnt结合[23]。此外,我们的研究表明,用Hedgehog信号中跨膜受体smoothened(smo)抑制剂索尼德吉(Sonidegib)靶向软骨生成功能障碍,恢复软骨细胞稳态,可显著阻止AS的骨融合进程[27]。作为Hedgehog通路的关键成分,特定的smo抑制剂也能够在血清转移模型的炎症后阶段预防新骨形成[41]。在自发的AS小鼠模型DBA/1中,过表达BMP拮抗剂noggin在预防和治疗策略上均有效,提示BMP可作为AS的潜在治疗靶点[42]。
3.6 肠道菌群调节剂胃肠道抑菌剂利福昔明可以调节肠道屏障,改善肠道菌群,抑制肠道炎症,并可以有效抑制AS进展[43]。相似地,吲哚-3-乙酸可以改变肠道菌群组成,减轻回肠组织病理性形态,并减缓了蛋白多糖诱导的AS动物模型的症状[44]。肠道菌群失调导致炎性小体活化以及IL-1β等炎性细胞因子的释放,而使用IL-1β阻滞剂阿那白滞素(anakinra)治疗AS的效果并不一致,但对于有高炎症指标或携带导致IL-1产生增加的变异基因 (如MEFVM694V变异)且对TNF抑制剂或IL-17抑制剂反应不足的患者,可以经验性地使用anakinra或其他长效IL-1拮抗剂进行治疗[11]。
4 总结与展望
AS是一种以骶髂关节炎引起的炎性背痛、外周关节痛以及关节结构性改变为特征的类风湿疾病,病程后期常出现脊柱融合现象。遗传因素、免疫因素和病理性成骨共同构成其发病机制。现有的治疗药物包括非甾体抗炎药、TNF抑制剂、靶向IL-23/IL-17的单克隆抗体以及缓解疾病症状的抗风湿药物。虽然这些药物能有效减轻活动期AS的炎症反应,但不能显著减缓AS的放射学进展和脊柱融合。对此合理的解释是,除了炎症环境外,AS中存在病理性异位成骨现象,包括近期揭示的软骨内成骨导致骨融合的崭新分子机制。因此,单纯的抗炎治疗不能减缓AS骨病变的发生,而抑制病理性成骨,如抑制破骨细胞活性、软骨形成、软骨内成骨、成骨细胞分化等,可能是治疗AS的一种有效的潜在方法[27-28]。
