反应增容结合立构复合结晶制备超韧PLA/PCL共混材料
2022-11-23杨皓然叶长春王佳贾光段同生刘双良高丽君崔静周立明方少明
杨皓然,叶长春,王佳,贾光,段同生,刘双良,高丽君,崔静,周立明,方少明
(1.郑州轻工业大学材料与化学工程学院河南省表界面科学重点实验室,郑州 450002;2.河南省中原塑料机械有限公司,郑州 450000)
随着工业的发展,各个产业对石油资源产品的需求逐年增大,导致石油资源被大量消耗并对环境产生了较大的影响,给社会的可持续发展带来巨大挑战[1-3]。为了减少对不可再生资源的依赖,开发环境友好型的新材料如生物基可降解的高分子材料越来越受到关注[4-5]。聚丙交酯[聚乳酸(PLA)]是一种生物基热塑性脂肪族聚酯材料,可完全生物降解,降解产物仅为二氧化碳和水,具有高强度、高模量和较好的生物相容性,常用于材料包装、纤维纺织和生物医学领域[6-7]。但是PLA固有的脆性大、韧性差的缺点严重限制了其应用领域,所以对PLA进行增韧改性成为PLA实际应用中的重要环节[8-9]。
聚己内酯(PCL)作为韧性可降解聚酯,加工性能优异,通常被用于增韧PLA[10-11]。由于PCL和PLA均含有端羧基、端羟基,可以与环氧基团、酸酐基团、异氰酸根等活性基团在熔融共混时原位反应生成酯基、醚键等新的化学键,因此可以采用如多元环氧低聚物(ADR)、赖氨酸二异氰酸酯(LDⅠ)等增容剂对PLA和PCL体系进行反应增容,从而提高两者的相容性[12-13]。然而,目前采用不同增容方法制备的PLA/PCL共混物韧性提高幅度较低,很难达到以弹性体作为增韧剂时的增韧效果,且PLA/PCL共混物很少能够同时具备较高的冲击韧性和拉伸韧性,因此还需要深入的研究,进一步提高PLA与PCL的界面结合,以充分发挥PCL的增韧效果。
近段时间,有研究人员发现了一种界面立构复合结晶(i-SC)的增容方法,即在反应增容的基础上,向左旋PLA(PLLA)/韧性聚合物共混体系中添加右旋PLA(PDLA),使共混体系在反应共混过程中原位形成位于PLLA和韧性聚合物界面处的立构复合晶体(SC晶体),进一步提高PLLA与增韧相的相容性,制备了超韧PLA共混材料[14]。笔者借鉴这一思路,同时考虑到异山梨醇基二异氰酸酯(ⅠBDⅠ)[15]作为一种生物基二异氰酸酯,具有合成所需原料非石油基且来源广泛、反应活性强等优点,因此选择其作为增容剂,尝试通过简单的两步加工方法,使共混体系在反应共混过程中原位形成位于PLLA和PCL两相界面处以及PLLA基体中的SC晶体,进一步提高PLLA与PCL的界面结合。为了控制SC晶体的分布,首先将PDLA,PCL和ⅠBDⅠ进行熔融共混(第一步),生成含有PDLA-ⅠBDⅠ-PCL分子的PCL/PDLA共混母粒,然后再将其与PLLA进行共混,制备PLLA/(PDLA/PCL/ⅠBDⅠ)共混物(第二步)。实验结果表明,采用上述方法制备的PLA/PCL共混物同时具备较高的缺口冲击强度和断裂伸长率,且其性能与ⅠBDⅠ的含量相关。同时,实验结果也证明了二异氰酸酯作为增容剂在i-SC增容路径中的有效性,相关结果对于实际生产具有指导意义。
1 实验部分
1.1 主要原材料
PLLA:4032D,美国Nature Works公司;
PDLA:D070,泰国道达尔科碧恩公司;
PCL:深圳光华伟业股份有限公司;
ⅠBDⅠ:自制。
1.2 主要设备及仪器
场发射扫描电子显微镜(FESEM):Regulus8100型,日本日立公司;
傅里叶变换红外光谱(FTⅠR)仪:TensorⅡ型,德国Bruker公司;
X射线 衍 射(XRD)仪:D8 Advance型,德 国Bruker公司;
差示扫描热量(DSC)仪:Q100型,美国TA公司;
电子天平:LCD-A1000型,赛多利斯科学仪器有限公司;
平板硫化机:QLB-50T型,江苏无锡市中凯橡塑机械有限公司;
万能试验机:UTM2000型,深圳三思纵横科技股份有限公司;
悬臂梁冲击试验机:S8225X型,上海斯玄检测设备有限公司;
真空干燥箱:DZF-6050AB型,北京中兴伟业仪器有限公司;
转矩流变仪:HaakePolyLabOs型,Thermo Scientic公司。
1.3 试样制备
为防止在混料时PLA含有水分,导致热降解影响制品的性能,在使用前需要将PLLA,PDLA材料放入真空干燥箱中,在80℃下干燥24 h。PCL材料则于50℃真空干燥箱中干燥24 h。ⅠBDⅠ需要低温、避光存储。
若要在PLLA与PCL熔融共混过程中原位形成位于两相界面处且与PCL通过化学键相连的SC晶体,共混体系中需要存在PDLA-PCL分子。由于ⅠBDⅠ上的异氰酸酯基与PLLA,PCL和PDLA上的端羟基的反应是随机的,因此反应共混时ⅠBDⅠ两端连接的分子种类不确定,若采用一步法将上述材料同时共混,无法保证一定能够生成PDLA-PCL嵌段共聚物。为了确保PDLA-PCL的生成,需要对混料顺序和步骤进行控制。首先利用ⅠBDⅠ与PDLA/PCL进行反应共混,得到含有PDLA-PCL分子的PDLA/PCL共混母粒(第一步);随后,将其与PLLA进行共混(第二步),母粒中的PDLA-PCL能够与PLLA结晶,原位形成位于PLLA和PCL相界面处的SC晶体。当然,由于第一步共混得到的母粒中还含有经ⅠBDⅠ扩链的PDLA-PDLA分子链和没有反应的PDLA分子链,因此第二步共混得到的材料中还存在位于PLLA基体中的SC晶体。实验具体共混工艺为:先将PDLA,PCL在转矩流变仪中熔融共混1 min后,加入不同含量的ⅠBDⅠ(添加量分别为0.5,1.0,1.5,2.0份),熔融共混9 min,得到含有PDLA-ⅠBDⅠ-PCL分 子 的 母 粒(简 称D/C/Ⅰx,x=0.5,1.0,1.5,2.0);然后将其与PLLA熔融共混6 min,得到PLLA/(PDLA/PCL/ⅠBDⅠx)(两步法)共混物[简称L/(D/C/Ⅰx)]。作 为对照,制备纯PLLA,PLLA/PCL,PLLA/PCL/ⅠBDⅠ(一步法)和PLLA/(PDLA/PCL)(两步法)共混物[分别简称L,L/C,L/C/Ⅰ和L/(D/C)]。上述共混物的熔融共混温度均为190℃,转速为60 r/min,配比见表1。
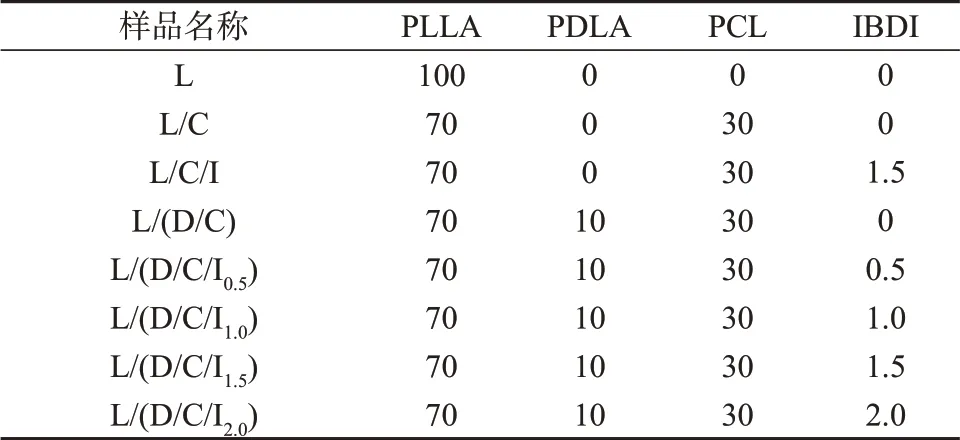
表1 共混材料各组分含量 份
性能测试样品制备:将共混物经预热(温度190℃、时间5 min)、热压(温度190℃、压力10 MPa、时间5 min)、冷压(温度25℃、压力10 MPa、时间5 min),依照GB/T 1843-2008制备缺口冲击样条和拉伸样条。
1.4 性能测试与结构表征
FTⅠR测试:测量范围500~4 000 cm-1,分辨率为4 cm-1,扫描次数为32次。
拉伸性能按GB/T 1040.1-2006进行测试,将样品竖直夹在夹具中间,以20 mm/min的速率拉伸样条直至断裂,数据采集频率为0.1 Hz,测试温度25℃,重复测试6次取平均值。试样尺寸为50 mm×4 mm×2 mm,原始标距为30 mm。
缺口冲击强度按GB/T 1843-2008进行测试,配合5.5 N冲击试验锤,测试温度25℃,重复测试5次取平均值。试样尺寸为80 mm×10 mm×4 mm,缺口为2 mm。
XRD测试:采用常温固定工作电压,扫描范围为5°~80°,步长为0.05°,停留时间为0.5 s。
DSC测试:利用DSC研究样品的热行为和结晶行为,温度扫描在氮气气氛下进行,样品质量为3~5 mg。测试熔融行为时,从25℃以10℃/min的速率加热到250℃;测试结晶行为时,从25℃以10℃/min的速率加热到190℃,保持5 min消除热历史,再以10℃/min的速率降温到25℃,保持5 min,最后以10℃/min的速率加热到250℃。
形貌表征:分别对样品冲击断面和液氮脆断面喷金60 s,在固定电压下利用FESEM观察样品形貌。
粒径分析:采用Nano Measurer软件对SEM照片中分散相尺寸进行统计,为克服SEM粒度分析法所存在的测定样品量太少、结果缺乏代表性的缺点,在实际操作时,对于每个样品测量200个颗粒以上求平均值。
2 结果与讨论
2.1 PDLA与PCL反应性分析
图1是D/C/Ⅰx共混物的转矩随时间变化曲线。从图1可知,在未加入ⅠBDⅠ之前,PDLA/PCL共混物的转矩随密炼时间的延长出现下降趋势,这是由于剪切变稀导致。加入ⅠBDⅠ后,D/C/Ⅰx体系的转矩随密炼时间的延长呈上升趋势,并且随ⅠBDⅠ含量的增加上升趋势更加明显,该结果表明ⅠBDⅠ和PDLA,PCL的端基发生反应生成了高分子量PDLAPDLA,PCL-PCL以及PDLA-PCL嵌段共聚物。

图1 D/C/Ⅰx共混物的转矩流变曲线
采用FTⅠR对ⅠBDⅠ与D/C/Ⅰx共混物进行了表征,如图2所示。由图2a可见,ⅠBDⅠ的N=C=O的伸缩振 动 峰 在2 274 cm-1处 清 晰 可 见,D/C/Ⅰx体 系 在2 274 cm-1处没有N=C=O基团的吸收峰,表明ⅠBDⅠ在密炼过程中已经完全反应。此外,对于D/C/Ⅰx体系,在1 606 cm-1处存在N—H的弯曲振动峰,在3 500 cm-1左右存在N—H的伸缩振动峰,这进一步表明PDLA和PCL分子链的端基与ⅠBDⅠ中的异氰酸酯基团发生了反应[16]。图2b中1 754 cm-1和1 724 cm-1处的吸收峰分别对应于PDLA和PCL的C=O基团的伸缩振动峰。需要注意的是,所有D/C/Ⅰx样品在测试前均经过索氏抽提将PDLA-PDLA,PCL-PCL以及没有反应的PDLA和PCL除掉。因此,FTⅠR和转矩流变测试数据可以共同表明第一步共混时ⅠBDⅠ与PDLA和PCL发生了反应,且反应物中形成了PDLA-PCL嵌段共聚物。
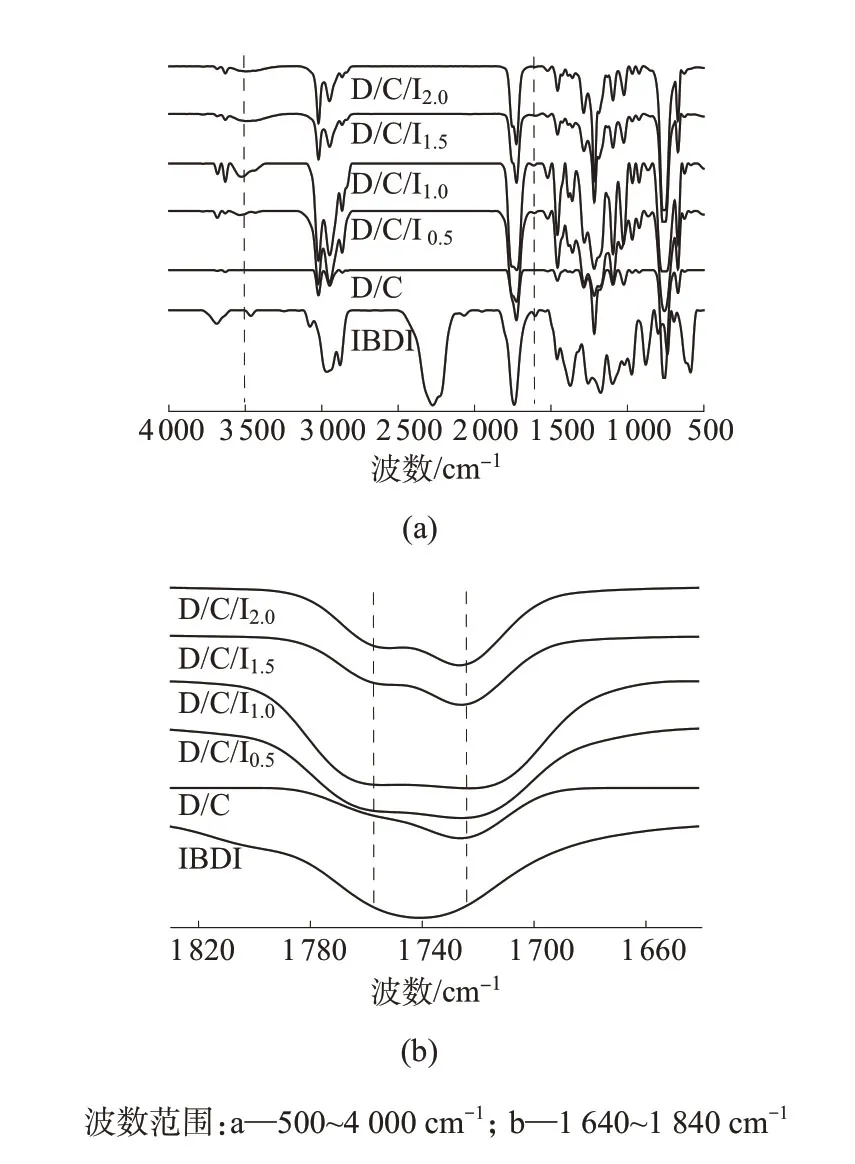
图2 ⅠBDⅠ和D/C/Ⅰx共混物的FTⅠR谱图
2.2 共混材料的结晶情况和结晶行为
图3 为纯PLLA及共混材料样品的XRD曲线。由图3可见,纯PLLA显示出一个宽的非晶衍射峰,这是由于PLLA结晶速度慢而成型过程中冷却速度快导致的。L/C和L/C/Ⅰ共混物在21.3°,22.0°和23.6°处观察到三个特征峰,其中21.3°和22.0°为PCL的(110)晶面,23.6°为PCL的(200)晶面,表明共混物中PCL处于结晶态。加入PDLA之后,L/(D/C/Ⅰx)体系分别在12.1°,20.8°,21.3°,22.0°和23.6°附近出现了5个明显的特征峰,12.1°和20.8°的峰分别对应于SC晶体的(110)和(300)/(030)晶面,其中的23.6°的特征峰既是PCL的(200)晶面又是SC晶体的(220)晶面。实验结果表明,PDLA的加入使样品中形成了SC晶体,该晶体可能位于PLLA和PCL两相界面处和PLLA基体中,由于前面的转矩流变和FT-ⅠR测试数据已经证明第一步共混后D/C/Ⅰx样品中有PDLA-PCL分子形成,所以必定有SC晶体存在于PLLA和PCL的相界面处。

图3 纯PLLA和共混材料的XRD曲线
表2为纯PLLA和共混材料样品的DSC统计数据,图4为纯PLLA和共混材料样品的DSC曲线。

图4 纯PLLA和共混材料的DSC曲线

表2 纯PLLA和共混材料的DSC升温(25℃升温至250℃)熔融数据
图4 a为纯PLLA及各组共混材料升温熔融曲线,用于表征压制成型样品的结晶情况。图4b和图4c分别为各样品由室温升温至190℃并保持5 min之后的降温曲线和二次升温曲线,用于表征各样品的非等温结晶行为。
图4a中,63℃附近出现的吸热峰归属于PCL结晶组分的熔融峰,在95℃附近出现的放热峰归结为样品中PLA非结晶组分在DSC升温测试过程中发生的冷结晶,168℃附近出现的吸热峰归属于PLA基体中α型均聚物晶体(HC晶体)的熔融峰,而在226℃附近出现的吸热峰归属于PLA基体中SC晶体的熔融峰。由表2可知,L的冷结晶峰温度为113.5℃,HC晶体的结晶度(Xc-HC)为9.3%,加入PCL后,冷结晶峰温度为93.2℃,Xc-HC增加至18.2%,这是由于加入PCL导致共混物黏度降低,PLLA分子链运动能力增强,有利于PLLA结晶。随着ⅠBDⅠ的加入,L/C/Ⅰ共混物的冷结晶峰温度转移到96.5℃,Xc-HC下降到15.3%,这是由于ⅠBDⅠ的加入使共混物中的链纠缠度增加所致。对于L/(D/C)样品,PDLA的加入使共混物的冷结晶焓降低,说明共混过程中产生的SC晶体能够起到成核剂的作用,在模压成型的冷却过程中促进PLA结晶,但由于冷却速度过快,基体并没有完全结晶,因此升温熔融曲线上仍有冷结晶峰的存在。对于L/(D/C/Ⅰx)体系,其Xc-HC均在7%左右,SC晶体的结晶度(Xc-SC)均在16%左右,说明有PDLA存在时,ⅠBDⅠ含量的变化对体系结晶行为影响不大。
从图4b和图4c可知,L/(D/C/Ⅰx)共混物在190℃降温到25℃的过程中在110℃附近出现结晶峰,再加热到250℃后,L(D/C/Ⅰx)共混物无冷结晶峰,这是由于L/(D/C/Ⅰx)体系中PLLA和PCL两相界面处和PLLA基体中存在SC晶体在190℃时没有熔融,其在以10℃/min的降温速率降温时可以起到成核点的作用,促进基体PLLA快速完成结晶,因此再次升温后没有出现冷结晶的现象。
2.3 共混材料的相形态
众所周知,材料的相形态是决定共混物力学性能的关键因素[17-18]。图5显示了各样品脆断面形貌的SEM照片。由图5可以看出,纯PLLA显示出光滑的断面。一般而言,两种不同材料混合在一起时,由于自身相差较大的属性,界面结合力弱,会导致聚合物之间发生相分离,少量的聚合物分散在基体中,形成海岛状的形态。可以看出L/C和L/C/Ⅰ共混物呈现出海岛结构,其中L/C共混物中PCL分散相颗粒粒径为3.3 μm,L/C/Ⅰ共混物中PCL分散相颗粒粒径为2.0 μm,粒径减小是由于ⅠBDⅠ的异氰酸酯基与聚合物的端基反应,使得PLLA和PCL界面相互作用得以改善,从而减小了粒径[19]。

图5 纯PLLA和共混材料的脆断面SEM照片
然而,当加入PDLA后,L/(D/C/Ⅰx)共混物中PCL相发生变化,呈不规则的形状。L/(D/C/Ⅰ0.5)和L/(D/C/Ⅰ1.0)共混物中PCL相明显发生变形,同样以不规则团状物形式存在,特别是在L/(D/C/Ⅰ1.5)和L/(D/C/Ⅰ2.0)共混物中观察不到明显的PCL团状物,且PCL分散相与基体PLLA相界面趋向于模糊,这说明D/C/Ⅰx和PLLA基体之间形成SC结晶,使得界面结合力大幅度提高[14,20]。作为对照,L/(D/C)共混物中PCL相也以团状物形式存在,但其与基体PLLA相界面较为明显并且部分聚集。
2.4 共混材料的力学性能
纯PLLA和共混材料的力学性能具体数据见表3。图6则为纯PLLA和共混材料的冲击断面SEM照片。由表3可以看出,纯PLLA在常温下呈现出硬而脆的特性,缺口冲击强度为2.5 kJ/m2,断裂伸长率为7%,拉伸强度接近70 MPa,断裂能为2.2 MPa。加入PCL之后,样品的缺口冲击强度提高至25.4 kJ/m2,断裂伸长率增加至27%,表明PCL能有效增韧PLLA。采用ⅠBDⅠ对共混材料增容后,L/C/Ⅰ的缺口冲击强度进一步提高至41.5 kJ/m2,但断裂伸长率变化不明显,说明单纯采用反应增容的方式无法使共混材料的缺口冲击强度和断裂伸长率同时提高。
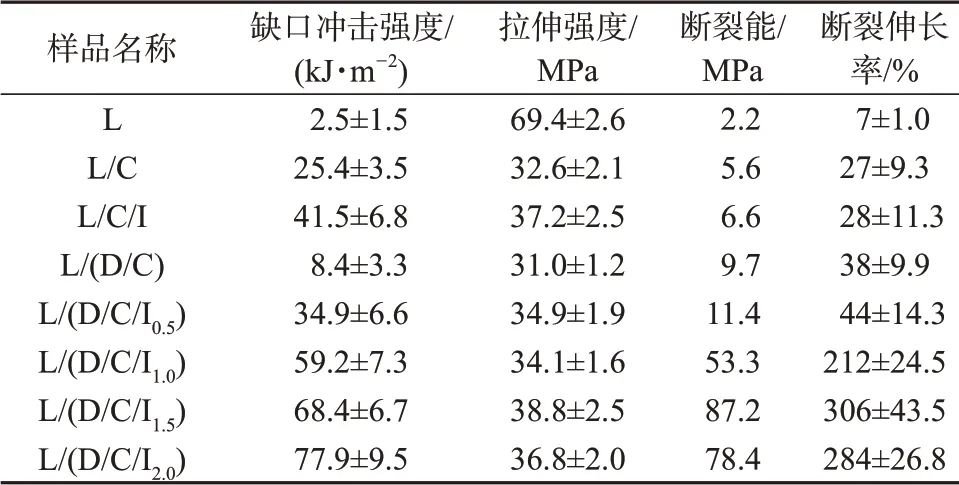
表3 纯PLLA与共混材料的力学性能
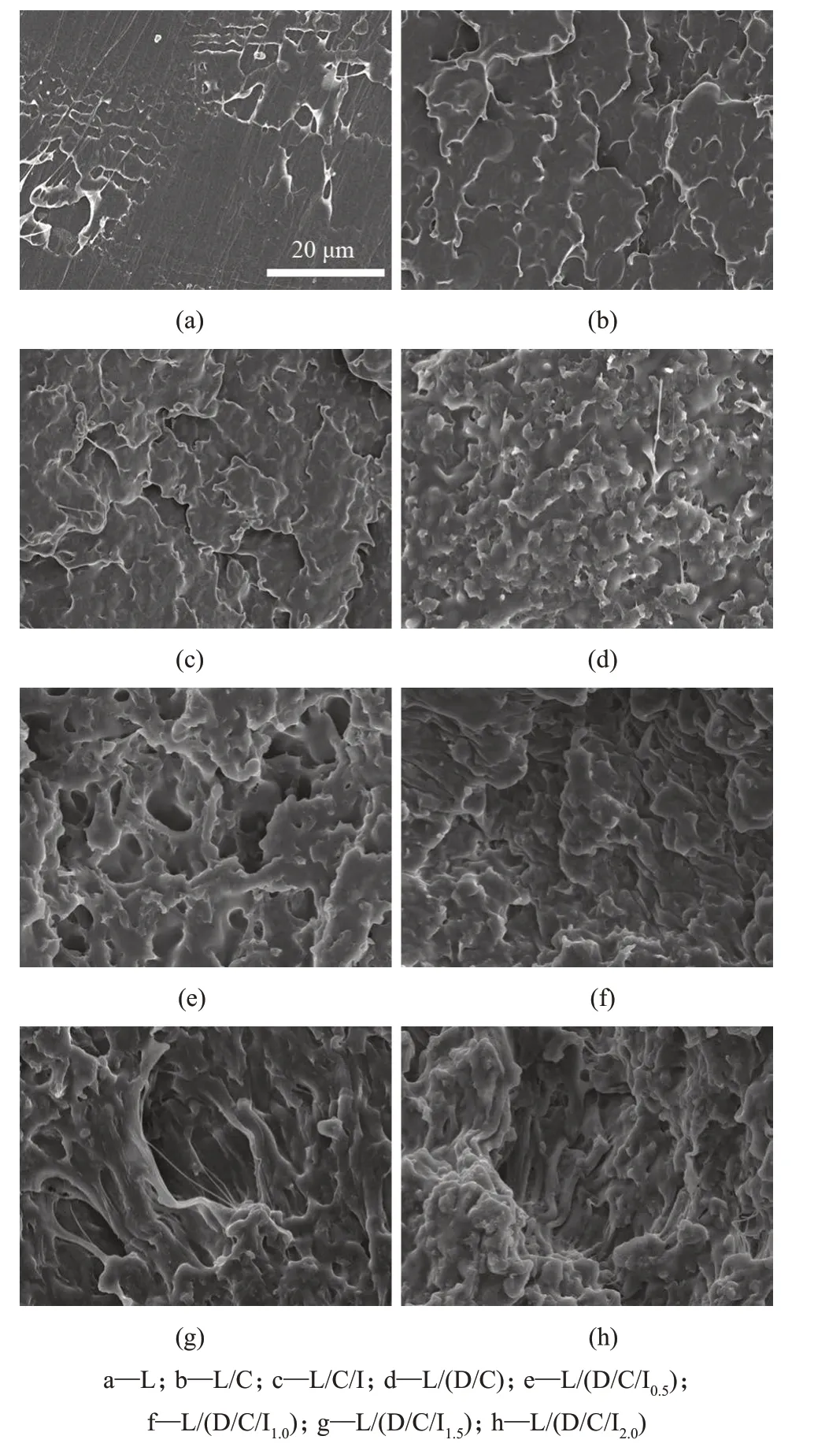
图6 纯PLLA与共混材料冲击断面的SEM照片
当PLLA/PCL共混体系中只加入PDLA时,L/(D/C)共混物的缺口冲击强度为8.4 kJ/m2,断裂伸长率为38%,拉伸强度为31 MPa,断裂能为9.7 MPa。加入0.5份的ⅠBDⅠ并采用两步法进行共混后,L/(D/C/Ⅰ0.5)共混物的缺口冲击强度提高至34.9 kJ/m2,但拉伸性能变化不大,当ⅠBDⅠ的含量为1份时,L(D/C/Ⅰ1.0)共混物的缺口冲击强度提高至59.2 kJ/m2,断裂伸长率大幅度提高至212%,继续增加ⅠBDⅠ的含量到1.5份,L/(D/C/Ⅰ1.5)共混物的缺口冲击强度提高至68.4 kJ/m2,断裂伸长率提高至306%、拉伸强度提高至38.8 MPa,断裂能提高至87.2 MPa,当ⅠBDⅠ添加至2份时,虽然L/(D/C/Ⅰ2.0)共混物的缺口冲击强度提高至77.9 kJ/m2,但拉伸性能有所下降。这些数据表明对于采用两步法反应共混制备的样品,由于两相界面处得到增强,其冲击韧性和拉伸韧性均得到大幅提高,采用该法成功制备了超韧PLA共混材料。
冲击断裂的表面形貌有助于了解材料的增韧机理[18]。由图6可以看出,PLLA共混物的断裂面光滑,呈现出脆性断裂;L/C共混物断裂面部分粗糙,并且在表面依旧可以观察到部分PCL颗粒;L/C/Ⅰ共混物的断裂面更加粗糙,观察不到PCL颗粒,缺口冲击强度达到41.5 kJ/m2,这是由于ⅠBDⅠ加入后相容性提高,使得样品宏观上冲击强度大幅提高。对于L/(D/C/Ⅰx)共混物,断裂表面都观察不到PCL颗粒,界面脱粘消失,整个断裂表面存在明显的基体塑性变形。其中L/(D/C/Ⅰ1.5)和L/(D/C/Ⅰ2.0)共混物的冲击断裂面出现明显的拉伸塑形形变,这表明界面粘附力的增强使得沿界面的传递更有效,从而在冲击断裂过程中消耗了更多的断裂能量。作为对照样品,L/(D/C)共混物的冲击断裂面呈现出粗糙的平坦面,没有观察到像L/(D/C/Ⅰx)共混物的塑形形变。
3 结论
以ⅠBDⅠ为增容剂、PCL为增韧相对PLLA进行增韧,通过PDLA的加入及混料顺序的控制,将反应增容与PLA的SC结晶相结合,成功制备冲击韧性和拉伸韧性同时提高的超韧PLA/PCL共混材料。结论如下:
(1)只进行反应增容时,L/C和L/C/Ⅰ共混物的分散相PCL都呈现出海岛结构,但L/C/Ⅰ的分散相粒径减小,韧性有所提高,这说明ⅠBDⅠ对PLLA和PCL有一定的增容效果。
(2)加入PDLA并采用两步法混料后,所有样品中均有SC晶体的形成。对于不加入ⅠBDⅠ的样品,L/(D/C)的分散相PCL尺寸较大,且韧性较差。但随着ⅠBDⅠ的加入及含量增加,L/(D/C/Ⅰx)共混物中的PCL分散相尺寸逐渐变小,且PCL分散相和基体PLLA的相界面趋向于模糊。该结果表明随着ⅠBDⅠ含量增加,PLLA和PCL界面处由于SC晶体的存在而结合力增强,增容效果提高。
(3)对于反应增容的两步法样品,随着ⅠBDⅠ含量的增加,相同含量PCL对PLLA的增韧效果大幅度提高:添加1.5份的ⅠBDⅠ时,样品缺口冲击强度为68.4 kJ/m2,断裂伸长率为306%;添加2.0份的ⅠBDⅠ时,样品缺口冲击强度为77.9 kJ/m2,断裂伸长率为284%。
