烟酰胺单核苷酸对酒精诱导大鼠肝细胞胰岛素抵抗的影响
2022-11-09郝丽萍
肖 琳,郝丽萍,罗 纲
(1.中南大学湘雅公共卫生学院营养与食品卫生学系,湖南 长沙 410078;2.华中科技大学同济医学院公共卫生学院营养与食品卫生学系,湖北 武汉 430030;中南大学湘雅公共卫生学院卫生毒理学系,湖南 长沙 410078)
胰岛素抵抗是贯穿Ⅱ型糖尿病发生发展全程的病理基础[1]。近年来研究表明,过量酒精摄入可能通过引起机体氧化损伤、炎症反应、脂调素代谢紊乱或胰岛素信号转导通路损伤等机制诱导外周组织发生胰岛素抵抗,而经典的胰岛素信号通路PI3K/Akt损伤是其中最为关键的机制之一[2-4]。沉默信息调节因子-1(silent information regulator 1,SIRT1)是一种氧化型烟酰胺腺嘌呤二核苷酸(oxidized nicotinamide adenine dinucleotide,NAD+)依赖的组蛋白去乙酰化酶,可通过调控PI3K/Akt信号通路的表达和激活来参与胰岛素信号转导[5-6]。我们前期研究显示,过量酒精代谢可通过降低大鼠肝脏内NAD+水平,进一步抑制SIRT1蛋白表达及下游PI3K-Akt通路磷酸化,阻滞胰岛素信号传导而最终诱导肝脏胰岛素抵抗,表明补充机体NAD+水平并进一步上调SIRT1蛋白表达可能是改善酒精诱导肝脏胰岛素抵抗状态的关键靶点[4]。
烟酰胺单核苷酸(nicotinamide mononucleotide,NMN)是一种自然存在的生物活性核苷酸,作为NAD+前体,其在机体内通过烟酰胺单核苷酸腺苷转移酶催化生成NAD+[7]。研究证实,口服NMN可完好无损的通过消化系统,体内的吸收非常迅速,3 min内进入血液,15 min内增加组织中NMN含量,并迅速提升血液、肝脏等器官中NAD+水平[8]。然而,NMN能否通过补充NAD+水平改善酒精诱导的肝脏胰岛素抵抗?潜在机制如何?目前尚未可知。因此,本文以前期研究为基础,从NAD+/SIRT1通路及下游PI3K-Akt通路探讨NMN改善酒精诱导肝脏胰岛素抵抗状态的相关机制。
1 材料
1.1 细胞原代大鼠肝细胞。
1.2 实验动物与试剂雄性SPF级SD大鼠5只,体质量(180~220)g,由中南大学实验动物学部提供。Williams’E培养基(W4125)、IV型胶原酶(C5138)及烟酰胺单核苷酸(纯度≥95%,货号N3501,批号Lot#SLBT9580)均购自于美国Sigma-Aldrich公司;I型鼠尾胶原(杭州欣友生物技术有限公司);胎牛血清FBS(美国Gibco);胰岛素(江苏万邦医药);无水乙醇(天津市科密欧试剂公司);Ex527(美国Selleck);葡萄糖检测试剂盒(北京中生北控生物科技有限公司);肝糖原检测试剂盒(A043-1-1,南京建成生物工程研究所);Cell Counting Kit-8(CCK-8)试剂盒(日本Dojindo);NAD+检测试剂盒(美国AAT Bioquest);RIPA裂解液(上海碧云天生物技术有限公司);Cocktail磷酸酶抑制剂(德国Roche);SIRT1抗体、p-PI3K抗体、PI3K抗体、p-Akt抗体、Akt抗体及HRP标记抗兔二抗购自于美国CST公司,ECL化学发光液及PVDF膜购自于美国Millipore公司。
1.3 仪器ELX800型酶标仪(美国BIOTEK),Western blot电泳仪(美国Bio-Rad),凝胶成像系统(美国Syngene),Master Flex 7524-40循环泵(德国Barnant)。
2 方法
2.1 原代大鼠肝细胞培养(胶原酶法)[9]大鼠隔夜禁食,消毒后背位固定于蜡板上。正中切口开腹,钝性分离并游离门静脉,插入留置针后固定。不含胶原酶的37 ℃预热灌流液以10~20 mL·min-1速度灌流,并剪断下腔静脉,等流出液体变清亮后,换含胶原酶的37 ℃预热灌流液继续灌流至肝脏变为土黄色且出现龟裂,停止灌流。分离肝脏至无菌平皿,用预冷培养基尽可能多吹散肝细胞,后用200目尼龙网进行过滤,4 ℃条件下50g低速离心5 min,重复3次获得较高纯度的肝细胞,接种于预铺鼠尾胶原的培养板,加入含10%FBS的Williams’E培养基,置于37 ℃、5%CO2条件下培养,用于后续实验。
2.2 建立酒精诱导大鼠肝细胞胰岛素抵抗模型根据本课题组前期对酒精诱导大鼠肝细胞胰岛素抵抗模型建立条件的摸索,原代大鼠肝细胞经含无水乙醇25 mmol·L-1的Williams’E培养基孵育培养24 h后,可发展为胰岛素抵抗模型。酒精作用时间、浓度及模型的稳定性在本课题前期研究中已进行验证[4]。
2.3 CCK-8实验检测NMN对原代大鼠肝细胞的毒性将原代大鼠肝细胞接种于96孔板,待细胞生长24 h后,分为空白组、阴性对照组和NMN组,空白组为无细胞的Williams’E培养基组,阴性对照组为有细胞的Williams’E培养基组,NMN组用Williams’E培养基配置终浓度为0.1、0.25、0.5和1 mmol·L-1溶液并分别加入对应培养孔(n=5)。培养24 h后加入含10 μL CCK试剂的100 μL培养基溶液,继续培养2 h后,于450 nm波长处测定吸光度值(OD),计算细胞存活率。
2.4 上清中葡萄糖消耗量检测(葡萄糖氧化酶法)细胞完成干预后收集细胞上清培养液,严格按照试剂盒说明书检测葡萄糖含量,以无细胞孔作为空白参照,计算细胞葡萄糖消耗量。
2.5 细胞内肝糖原合成量检测收集已完成干预细胞,采用蒽酮法检测细胞内肝糖原含量,详细实验操作参照试剂盒说明书进行。
2.6 肝细胞NAD+含量检测收集已完成干预细胞,使用试剂盒内裂解液裂解30 min后,在4 ℃条件下转速5 000 r·min-1离心5 min,分离上清液测定NAD+含量,具体实验操作参照试剂盒说明书进行。
2.7 Western blot检测SIRT1、p-PI3K/PI3K、p-Akt/Akt含量收集已完成干预细胞,用含PMSF及磷酸酶抑制剂的RIPA裂解液提取总蛋白,BCA蛋白定量并调节各样品总蛋白浓度一致。用微量上样针加样及Marker,80 V稳压电泳,完成后湿法转膜,将蛋白转移至已活化的PVDF膜上,再放入5%的脱脂奶粉封闭液内封闭1 h。洗脱缓冲液洗膜3次,加入稀释比为1 ∶1 000的一抗4 ℃过夜,再洗膜3次,加入稀释比为1 ∶5 000的二抗孵育1 h,最后洗膜3次,进行显影,获得图像使用Quantity One软件进行分析。

3 结果
3.1 不同浓度NMN对原代大鼠肝细胞活性影响如Fig 1所示,在0~1 mmol·L-1的范围内,NMN对原代大鼠肝细胞存活率无明显影响,参照已有研究使用的有效浓度[10],后续选择0.1 mmol·L-1及0.5 mmol·L-1浓度进行实验。
3.2 不同浓度NMN对酒精诱导的肝细胞胰岛素抵抗模型葡萄糖消耗量及肝糖原含量的影响根据选择的NMN干预浓度设置5个分组,分别为:对照组(Con)、酒精模型组(Eth)、酒精+低浓度NMN组(Eth+LNMN,NMN浓度为0.1 mmol·L-1)、酒精+高浓度NMN组(Eth+HNMN,NMN浓度为0.5 mmol·L-1)、NMN对照组(NMN,NMN浓度为0.5 mmol·L-1),干预24 h后检测指标。结果如Tab 1所示,与对照组相比,酒精模型组的葡萄糖利用率及肝糖原含量均明显下降(P<0.05),出现明显的胰岛素抵抗状态。相反,与酒精模型组相比,高、低浓度的NMN干预均可明显升高细胞的葡萄糖利用率及肝糖原含量(P<0.05),表明NMN具有改善酒精诱导的肝细胞胰岛素抵抗的功能。此外,NMN单独干预对肝细胞的葡萄糖利用率及糖原含量无明显影响。

Tab 1 Effects of ethanol and NMN on glucose consumption and glycogen content in primary rat hepatocytes
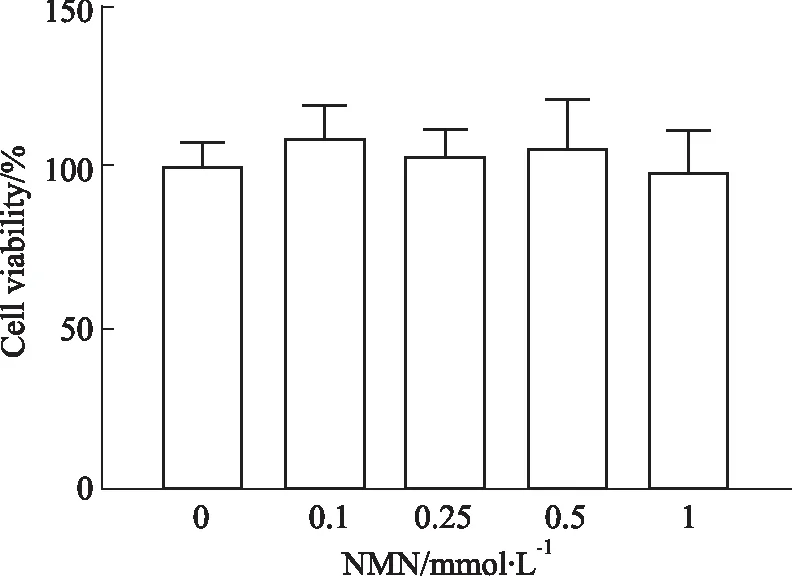
Fig 1 Effects of NMN on cell viability of primary rat hepatocytes
3.3 不同浓度NMN对酒精诱导的肝细胞胰岛素抵抗模型中PI3K-Akt信号通路的影响PI3K-Akt通路磷酸化是胰岛素信号转导不可缺少的关键步骤[5]。如Fig 2所示,与对照组相比,酒精暴露明显降低细胞内PI3K及Akt的磷酸化水平(P<0.01),阻滞胰岛素信号转导。而与酒精模型组相比,高、低浓度的NMN则可明显增加PI3K及Akt的磷酸化水平(P<0.05),恢复胰岛素信号通路正常转导,表明NMN改善酒精诱导的肝细胞胰岛素抵抗与调节PI3K-Akt信号通路活化密切相关。
3.4 不同浓度NMN对酒精诱导的肝细胞胰岛素抵抗模型中NAD+含量及SIRT1蛋白表达影响NAD+依赖的去乙酰化酶SIRT1是PI3K-Akt信号通路的上游调节蛋白[5],酒精在肝细胞内代谢过程伴随NAD+向NADH大量转化[4]。如Fig 3所示,酒精暴露明显降低了细胞内NAD+含量(P<0.05)并抑制SIRT1蛋白表达(P<0.05)。与预期一致的是,高、低浓度的NMN干预均有效补充了细胞内NAD+水平(P<0.01,P<0.05),同时明显提高SIRT1蛋白表达水平(P<0.05),提示NMN极有可能是通过恢复NAD+/SIRT1通路水平,进一步调控PI3K-Akt信号通路改善酒精诱导的胰岛素抵抗。
3.5 SIRT1抑制剂Ex527对NMN改善酒精诱导的肝细胞胰岛素抵抗作用的影响为了进一步验证NAD+/SIRT1通路在NMN改善酒精诱导的肝细胞胰岛素抵抗过程中的关键作用,我们利用SIRT1的抑制剂Ex527进行验证。设置4组,分别为:对照组(Con)、酒精模型组(Eth)、酒精+NMN组(Eth+NMN,NMN:0.5 mmol·L-1)和酒精+NMN+Ex527组(Eth+NMN+Ex527,Ex527:10 μmol·L-1)。如Tab 2和Fig 4,与Eth+NMN组相比,Eth+NMN+Ex527组的葡萄糖利用率及肝糖原含量明显下降(P<0.05),p-PI3K及p-Akt水平明显降低(P<
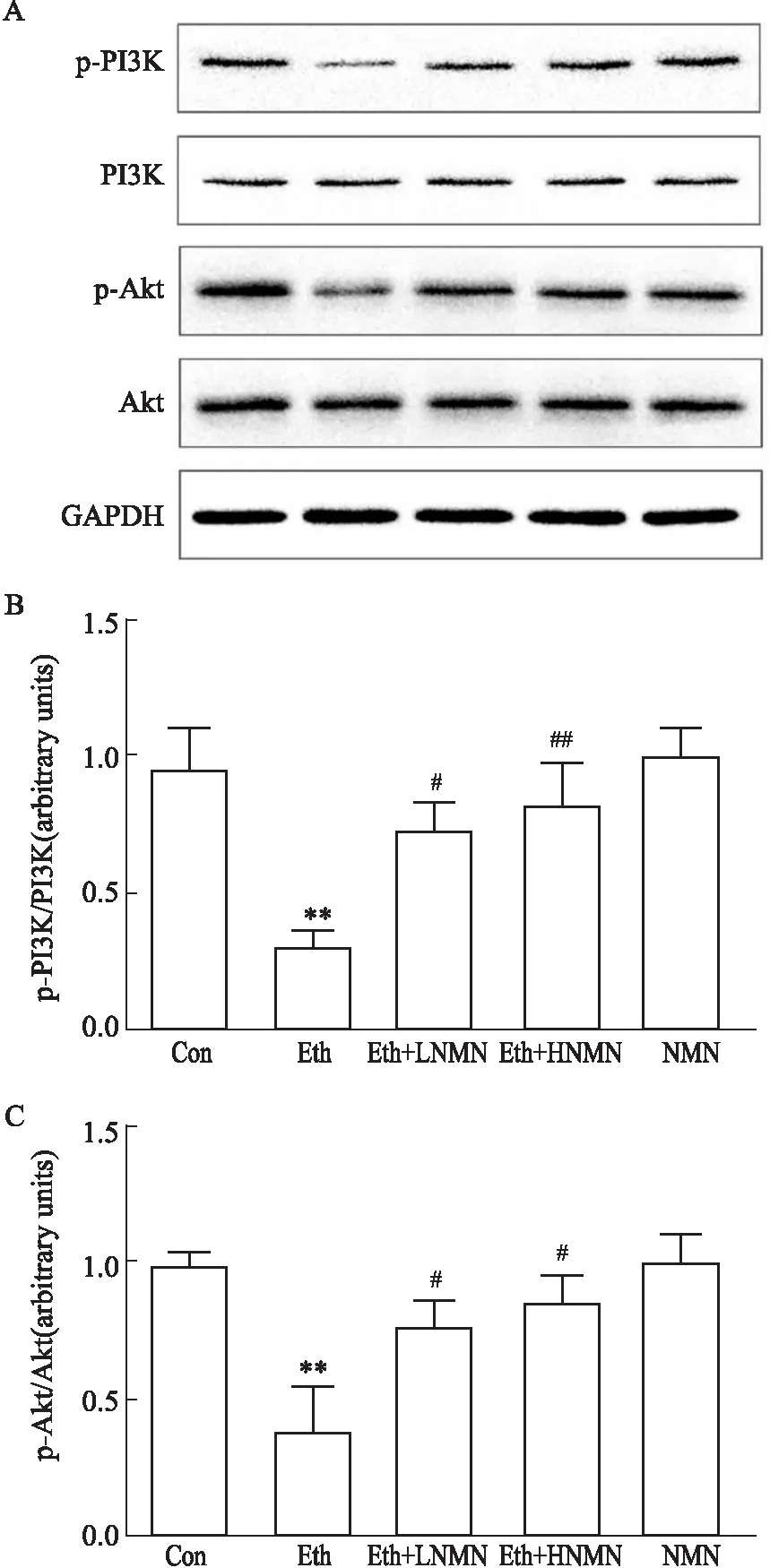
Fig 2 Effects of ethanol and NMN on insulin signaling pathway

Tab 2 Effects of Ex527 on glucose consumption and glycogen content in primary rat hepatocytes
0.01),但NAD+水平无明显变化,表明NMN对酒精诱导肝细胞胰岛素抵抗的改善效应可被Ex527抑制。

Fig 3 Effects of ethanol and NMN on NAD+ content and SIRT1 expression in primary rat hepatocytes n=4)
4 讨论
胰岛素信号转导通路损伤是胰岛素抵抗发生的核心机制之一。在胰岛素抵抗状态下,肝细胞葡萄糖利用率及糖原含量将明显降低[11-12]。已有多项研究表明,酒精可能通过抑制PI3K-Akt通路活化而诱导肝细胞、肌细胞、脂肪细胞等多种细胞发生胰岛素抵抗[2,4,13]。相反,通过有效干预恢复PI3K-Akt通路活化则能明显改善细胞胰岛素抵抗状态[14-15]。在本研究中,酒精暴露可诱导原代大鼠肝细胞发生胰岛素抵抗,NMN干预则明显增加了肝细胞葡萄糖利用率及糖原含量,表明NMN对酒精诱导的肝细胞胰岛素抵抗状态具有良好的改善效应。此外,Western blot结果显示,NMN能够明显增加PI3K-Akt通路的磷酸化水平,提示这种改善效应与NMN恢复PI3K-Akt胰岛素信号通路功能密切相关。然而,NMN如何调控PI3K-Akt通路,仍未可知。

Fig 4 Ex527 antagonized protective effects of NMN on ethanol-induced hepatic insulin
NAD+是细胞生命活动中参与多种生物学进程必不可少的辅酶。研究显示,补充NAD+水平对包括肥胖、非酒精性脂肪肝、糖尿病在内的多种代谢性疾病有良好的治疗效应[7,16]。值得注意的是,由酒精在机体内代谢引发的整体NAD+水平降低被认为是酒精造成多种细胞损伤的关键始动环节[4,17]。我们前期研究也显示,酒精阻滞肝细胞PI3K-Akt胰岛素信号通路与其抑制NAD+/SIRT1通路密切相关,以上研究共同提示恢复NAD+/SIRT1通路极有可能是改善酒精诱导肝细胞胰岛素抵抗的有效手段。然而,针对此靶点的研究仍处于起步阶段,极为有限的研究显示另一种NAD+前体烟酰胺核糖可通过补充肝组织NAD+水平改善酒精引发的小鼠肝损伤[18]。此外,白藜芦醇可能通过调控酒精代谢过程,补充NAD+水平以改善酒精诱导的胰岛素抵抗[4],而相较于白藜芦醇,NMN作为NAD+前体,具有更为直接补充机体NAD+水平的效力。在本研究中,酒精暴露可降低原代大鼠肝细胞的NAD+水平及SIRT1表达量,而NMN干预可明显恢复NAD+水平、SIRT1表达量以及PI3K-Akt的磷酸化水平;相反,在SIRT1的抑制剂Ex527存在的情况下,尽管NMN有效恢复了酒精降低的NAD+水平,Ex527仍然阻断了NMN上调PI3K-Akt磷酸化水平的作用,以上结果均表明,NMN可通过上调NAD+/SIRT1通路,进一步调控PI3K-Akt通路活化而改善酒精诱导的肝脏胰岛素抵抗。
综上所述,NMN具有改善酒精诱导肝脏胰岛素抵抗的作用,其机制与NMN恢复NAD+/SIRT1通路功能,并进一步活化胰岛素信号通路PI3K-Akt相关,这不仅为NMN干预防控酒精诱导2型糖尿病提供了依据,同时也为NMN干预防控更多的酒精相关疾病提供了机制参考。
