成年商品猪肠道可培养核心菌群的结构与组成分析
2022-11-08蔡淳理杨大容刘宝生
田 野,蔡淳理,杨大容,李 慧,刘宝生,3*
(1.江西农业大学 动物科学技术学院,江西 南昌 330045;2.四川农业大学 动物医学院,四川 成都 611130;3.江西农业大学 兽药研究所,江西 南昌 330045)
动物肠道作为微生物最密集的栖息地,长期寄居着大约1014的微生物个体[1]。单胃哺乳动物胃肠道微生物主要是由细菌组成,约占机体微生物总量的78%[2]。据统计,肠道菌群中含有500~1 000种细菌[3],其中已被记录的菌门有50多个[4]。肠道菌群结构并非固定,但在哺乳动物肠道中通常是以Bacteroidetes、Firmicutes中的1个或2个门占据优势[5]。动物肠道菌群与年龄有关,有研究表明,仔猪肠道菌群随着发育过程逐渐转化并趋于稳定[6],而成年猪的肠道菌群大致稳定,总体上以Proteobacteria、Firmicutes、Bacteroidetes为主[7-8]。
近十多年来,因为高通量测序技术的飞速发展,虽然在猪肠道菌群的结构、功能,以及肠道菌群与宿主关系等多方面展开了丰富的研究[9-10],但迄今为止肠道内生菌群的真实情况仍旧不够明朗。16S rRNA基因全长1 540 bp,包含9个可变区,是目前细菌种属鉴定和分类中应用最广泛的“biomarker”[11-12]。目前的高通量测序技术受制于本身测序长度的限制,往往只能选择1~3个可变区作为靶点[13]。Claesson等[14]通过比较16S rRNA基因的V3+V4和V4+V5两组高变区在Roche-454和Illumina 2个主流测序平台中对细菌种属特征分析的偏差,发现基因序列的覆盖率越大,准确性越高。为了避免高通量测序中的有限测序在菌群多样性评价和估计上的不足,本研究通过基于16S rRNA基因的全序列扩增与测序,构建成年商品猪肠道普通可培养核心菌群(general culturable core bacteria, GCCB)的16S rRNA基因克隆文库,利用Mothur、bioEdit、DNAstar等生物学软件,对猪肠道内源GCCB的多样性进行分析,为更好地阐明猪肠道不同肠段的菌群多样性及其分布特征提供参考。
1 材料与方法
1.1 肠道GCCB的分离与筛选
肠道菌群的分离方法同李慧等[15]。简言之,随机选择3头成年健康商品猪,分3次分别独立采样。按猪肠道的延续方向,自前向后将十二指肠、空肠、回肠、盲肠、结肠和直肠依次分成3、5、1、2、3、3个节段,无菌操作,分别从每一节段中采集肠道内容物,利用PCA(plate count agar)、NA(nutrient agar)、TSA(tryptic soy agar)、LB 营养琼脂(Luria-Bertani nutrient agar)和标准Ⅰ号琼脂(standard I nutrient agar,StdI)5种实验室常用非选择性培养基(见表1)分别对每一肠段的肠道菌群进行10倍梯度分离培养。根据不同培养基、不同肠段所分离菌群菌落形态的不同,选取每一肠段在每种不同培养基中数量最多、形态典型的3种不同类型菌落(如果菌落形态单一,选取的菌落数可小于3)组成该肠段的GCCB。考虑到同一肠段中的同种优势菌种可能在五种培养基中反复出现,导致GCCB中同种菌种的重复采集,在目测菌落形态相同或相似的情况下,同一肠段不同培养基之间,相同的菌种只选择其中1株作为GCCB成员。所有纳入GCCB的菌株经过进一步纯化培养后,挑单菌落接种至各自分离平板对应的液体培养基中,过夜培养后,制备成甘油冻干管,保藏在-40 ℃冰箱中备用。

表1 5种菌群分离用非选择性培养基的配方
1.2 核心菌株的活化与培养
从-40 ℃冰箱中取出事先保存好的肠道核心菌株甘油管,自然解冻。无菌操作,将其接种至事先灭菌好的空白TSB(或NB)液,盖紧后置于37 ℃、120 r/min摇床中培养12~24 h,使冷冻菌株复苏。取复苏菌液划线接种TSA(或NA)平板,置于37 ℃培养箱中培养18~24 h,用于确认菌株的纯一性。确认纯一的菌株则按2%的接种量用空白TSB(或NB)液连续传代活化2次,取活化好的过夜培养液,用于16S rDNA的扩增或细菌全基因组的提取。
1.3 16S rDNA序列的扩增
取1 mL新鲜活化好的核心菌株培养液,10 000 r/min离心5 min,弃上清后用1 mL双蒸水洗涤沉淀菌体细胞1次,同条件再次离心弃上清,沉淀菌体细胞用1 mL双蒸水重悬后即作为DNA模板加入25 uL PCR反应体系(无菌ddH2O、2×SanTaq PCR Mix(上海生工:B532061)、上下游引物),利用引物(27F:5′-AGAGTTTGATCCTGGCTCAG-3′;1492R:5′-ACGGTTACCTTGTTACGACTT-3′)进行PCR扩增(95 ℃ 5 min;95 ℃ 30 s,56 ℃ 30 s,72 ℃ 90 s,25个循环;72 ℃ 8 min)。阴性对照用无菌ddH2O替代DNA模板进行。对于少量直接用菌体细胞重悬液作为DNA模板无法扩增出其16S rDNA的菌株,则利用细菌基因组DNA快速抽提试剂盒(上海生工:B518225)先进行基因组的提取(提取方法参照试剂盒操作说明书),然后再用提取后的基因组DNA作为16S rDNA序列扩增的模板按上述方法进行PCR扩增。
所有PCR产物均用1%的琼脂糖凝胶电泳(200 V,30 min),进行纯度与浓度的检测,取条带单一且清晰、亮度足够,位于DNA Marker 所示1.5 kb左右的样品,送往擎科生物(武汉公司)进行测序。
1.4 16S rDNA测序
所有样品均采用一代Sanger法进行序列测定,为了测出16S rDNA扩增产物的全序列,本研究采用双向测通方案进行测序。测序结束后,由测序公司负责将双侧序列拼接成一条完整的16S rDNA序列。
1.5 GCCB的OTU聚类分析
将测序获得全部菌株的原始16S rDNA序列,根据数据处理的相关要求,使用Mothur、bioEdit、DNAstar等生物学软件,对原始序列进行质检、筛选、校正、对齐、合并等处理。然后经Mothur软件的summary程序检测,去除前后引物并校正后的序列平均长度在1 400 bp左右。针对16S rDNA全序列测序结果,采用去冗余、嵌合体检测、多重比对、过滤、双序列距离矩阵比对等程序处理,然后根据furthest neighbor算法,选择Mothur软件对遗传信息距离矩阵进行聚类。将97%以上相似度的序列归类并作为一个OTU(operational taxonomic unit)。提取OTU代表性序列文件,使用MEGA 6.0软件进行Clustal W算法比对,然后采用Neighbor-Joining算法构建并绘制进化树。
1.6 GCCB的NCBI在线BLAST分析
登录NCBI BLAST网址(https://blast.ncbi.nlm.nih.gov/Blast.cgi)进行在线分析。在nucleotide数据库中采用 megablast法对所有序列进行逐一比对。根据E值(Expect)、一致性(Indentities)、插入或缺失(Gaps)3个主要参数进行结果分析判别。按照相似性好、匹配度高、Gaps少的标准来确认每条序列最佳匹配物种序列。在门(phylum)、目(order)、科(family)、属(genus)4个分类学水平上分析总结各肠道、各节段的菌群组成和分布特征。
1.7 GCCB的多样性分析
基于NCBI BLAST分析各肠道菌群结构的结果,使用Mothur软件进行α多样性分析并计算菌群多样性指数。在多样性方面,Shannon 指数反映了群落的多样性,Simpson 指数反映了群落中优势物种的集中程度;在丰度方面,Chao1 指数和ACE指数对群落中的稀有菌种具有更好的估计。Shannon 指数、Chao1 指数、ACE指数越大,Simpson 指数越小,则样品中的物种多样性越高。
1.8 GCCB在不同肠段的分布特征分析
综合NCBI在线BLAST的分析结果及Mothur的OTU聚类分析数据,对猪肠道中的可培养核心菌群结构进行归类处理。估计猪肠道可培养核心菌群在各个肠段的OTU真实分布,绘制不同OTU的菌株数在不同肠段的分布热图。根据全部肠道不同节段的菌株分布,利用heatmap软件(“http://www.heatmapper.ca/”,Heml软件)在种水平上绘制heatmap图。同时归纳并描述各肠段分离可培养核心菌群的交叉分布信息,绘制各肠段菌群分布的Venn图(http://jvenn.toulouse.inra.fr/app/example.html)。
1.9 相关数据的统计学分析
可培养核心菌群的组成以及其多样性指数等的统计学数据处理,使用SPSS 24.0软件进行相关性分析和ANOVA分析,P<0.05表示差异显著。
2 结果与分析
本研究共对分离自健康成年商品猪6个肠段17个肠节段的共355株可培养核心菌株进行了16S rDNA全序列的检测,其中十二指肠83株、空肠84株、回肠30株、盲肠48株、结肠58株、直肠52株。
2.1 OTU聚类与菌群结构分析
在97%的相似水平上对全部核心菌株的16S rDNA进行聚类分析,共获得32个OTU,物种注释结果显示它们分属于8个菌属,17个菌种(见表2)。统计分析结果表明,各肠段分离的核心菌株数量与OTU的聚类个数无显著相关性(P>0.05,见图1)。

图1 不同肠段GCCB的OTU聚类分析结果
将聚类所得32个OTU用MEGA 6.0软件进一步绘制种属进化树(见图2),将所得进化树结合物种注释结果(见表2)来看,同一菌属下的种、亚种均能较好的聚集到1个分支上,各分支间的距离也能在一定程度上体现种属间的差异水平,展示物种之间的亲缘关系。

图2 核心菌群OTU聚类后的种属进化树绘制

表2 成年猪肠道GCCB的OTU聚类分析结果
2.2 肠道可培养核心菌群16S rDNA的NCBI在线BLAST结果与分析
本研究同时对所有核心菌株的16S rDNA序列进行NCBI在线数据库的逐一比对。将比对结果按种属结构进行统计,355株核心菌株分别属于3个菌门、5个菌目、6个菌科、13个菌属(见表3)、33个菌种及亚种。核心菌群中以Escherichia、Bacillus、Proteus3个菌属丰度最高,分别占据检测样本的41%、30%和9%。

表3 成年猪肠道GCCB的结构组成
2.3 α多样性分析
α多样性的分析结果(表4)表明,成年商品猪肠道中,可培养核心菌群多样性最丰富的为结肠和空肠,其次为直肠和十二指肠,多样性最简单的则为回肠和盲肠。

表4 猪肠道GCCB的α多样性指数分析
2.4 成年猪不同肠段GCCB的结构及其分布特征分析
从本研究的结果来看,355株GCCB在门水平上,除其中1株为Actinobacteria外,其余均为Proteobacteria和Firmicutes。在全部检出的33个菌(亚)种中,小肠共检出27种,大肠共检出26种,其中大、小肠共有菌种17种。不同肠段间,十二指肠共检出8个菌属,16个菌种,其中丰度排列前3的菌种依次为Escherichiacoli、Bacillusamyloliquefaciens和Bacillusvelezensis;空肠共检出9个菌属,20个菌种,其中丰度最高的3种为Escherichiacoli、Bacillusvelezensis和Bacillussubtilis;回肠共检出6个菌属,10个菌种,丰度排列前3的菌种为Escherichiacoli、Cronobactersakazakii和Proteusmitabilis;盲肠共检出6个菌属,10个菌种,丰度最高的前3菌种为Escherichiacoli、Bacillusvelezensis和Proteusmitabilis;结肠共检出10个菌属,20个菌种,丰度最高的前3菌种为Escherichiacoli、Klebsiellaquasipneumoniae和Bacillusvelezensis;直肠共检出8个菌属,15个菌种,丰度排列前3的菌种为Escherichiacoli、Cronobactersakazakii、Bacillusvelezensis(见图3)。

图3 成年商品猪不同肠段的GCCB组成结构图
通过对不同肠段中检出菌种的相对丰度分析(图4),发现Escherichiacoli在所有肠段均有检出,且在各肠段中均是检出数量最多的菌种,其中盲肠是检出概率最高的肠段,占全部检出菌株的56.25%。Bacillusvelezensis在盲肠、结肠、直肠,以及十二指肠、空肠均是主要检出菌种,但在回肠却没有检出。Bacillusamyloliquefaciens和Proteusmirabilis在各肠段均有分布,其中前者以十二指肠的检出频率最高,后者则在回肠中的相对含量更高。此外,Cronobactersakazakii虽然在十二指肠、盲肠和结肠都没有检出,但它在回肠中的检出频率却仅次于Escherichiacoli。
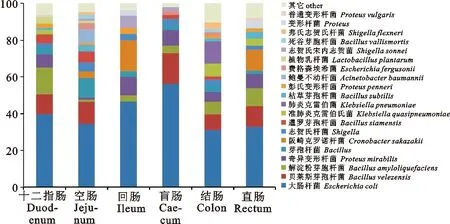
图4 成年商品猪不同肠段GCCB相对丰度图
为了进一步分析各肠段间共有菌群的分布特性,本研究还对全部检出菌种进行了分类统计,结果显示,小肠、大肠各肠段之间的共有菌种均为6种,而在整个肠道的六个肠段均有分布的菌种有5种(图5),分别是Escherichiacoli、Bacillusamyloliquefaciens、Shigella、Bacillus和Proteusmirabilis,占全部检出菌总数的60.56%。Proteuspenneri在小肠中广泛分布而在大肠中仅在结肠中检出,同样,在大肠中普遍存在的Bacillusvelezensis在回肠中也没有发现。

图5 不同肠段共有核心菌群的分析
同时,本研究还对同一肠段不同肠节段之间的可培养核心菌群进行了比较分析。从图6可以看出,Escherichiacoli虽然广泛分布于整个肠道,但检出率最高的肠段还是回肠、盲肠和十二指肠前段。Cronobactersakazakii虽然不是在肠道的每一节段都有检出,但其在回肠和直肠前段的检出率却很高。Bacillusamyloliquefaciens广泛分布于十二指肠、回肠和大肠各段,但在空肠各节段中却很少检出。类似的还有Bacillusvelezensis,该菌广泛分布在肠道的大多数节段,但回肠和直肠前段却没有检出。

图6 猪肠道不同节段可培养菌群的分布特征
3 讨 论
3.1 成年猪肠道GCCB的总体结构特征
本研究利用5种不同的非选择性培养基,对商品猪不同肠段不同节段的GCCB进行了分离培养,通过对分离菌株16S rDNA全序列的检测与分析,发现成年商品猪肠道内的GCCB绝大部分均属于Proteobacteria和Firmicutes 2门,而且以Escherichia、Bacillus、Proteus3个属为主,占样本检出量的80%。虽然本研究检测的只是肠道中的GCCB,但从检测结果来看,肠道菌群的总体构成与Kelly等[16]和Zhao等[17]通过高通量的方法对不同肠段内菌群多样性的研究结果相一致,而与Regina等[18]和Kim等[19]利用粪便样品所测得的猪肠道菌群主要由Firmicutes和Bacteroidetes构成的结果不完全相同。因此可以推断,动物肠道内与动物粪便的菌群结构是有差异的,对动物肠道菌群的研究来说,仅仅检测粪便样品中的菌群结构是不够的。
3.2 成年猪肠道GCCB不同肠段的结构特征
本研究因为菌群分离时不同肠段内容物的稀释倍数不同,因此不同肠段分离所得的菌株在各肠段的实际数量也是不同的,但所有分离菌株均为所在肠段含量最多的可培养菌种,因此能很好地反映不同肠段和节段中的可培养优势菌群组合。从不同肠段GCCB的结构来看,Escherichiacoli、Bacillusamyloliquefaciens、Shigella、Bacillus和Proteusmirabilis5种菌是广泛分布于所有肠段的共有菌。同时本研究还发现尽管空肠的可培养菌含量相对不高[20],但其种类却比较丰富,而盲肠含菌量相对丰富,但其菌群的多样性却不高。Looft等[21]认为,不同肠段中菌群结构的不同是基于不同菌群在肠道中所起的作用不同,小肠菌群多与肠道内小分子物质的吸收有关,而大肠菌群则与肠道内容物中的植物细胞壁降解有关。同样,Kelly等[16]认为不同肠段的菌群构成还因其肠道粘膜毛细血管扩散出来的氧分及肠腔内容物中的营养成分的含量不同而不同,十二指肠和空肠粘膜菌群绝大部分为Proteobacteria,随着肠道向后延续,在回肠和大肠部分Proteobacteria逐渐减少,而Bacteroidetes和Firmicutes依次增多。由于菌群分离过程的工作量比较大,本试验未同时对成年猪肠道中的厌氧可培养菌群进行分离筛选,因此,本研究的结果仅代表普通可培养核心菌群的结果。
3.3 肠道菌群多样性分析中16S rDNA全长测序与局部测序的差异
在当前动物肠道菌群的结构与多样性研究中,16S rDNA的高通量测序分析法已成主流,但由于测序技术所限,目前大多都是基于16S rDNA的1个或2个可变区进行,无法覆盖整个16S rRNA基因,因此必然存在比对信息不足、比对结果准确度偏低等问题[22-23]。为了避免此类问题的发生,本试验采用了16S rDNA的全序列进行测序并比对。通过Mothur软件的OTU聚类分析,结果表明本研究所分离菌株分别属于8个属,17个种。然而,通过16S rDNA全序列的NCBI在线比对结果却显示本研究所分离菌株分为13个属,33个种或亚种。这一结果表明,OTU的聚类分析虽然具有很好的科学依据,但它作为一种纯粹的计算机算法,依然存在一定系统缺陷[13],某些预测的OTU并不能代表真实的细菌种类,并与传统的菌群分类学分类不符[24]。
4 结 论
成年商品猪肠道中的GCCB主要包括Escherichia、Bacillus、Proteus3个菌属中的十几个菌种,在这些菌种中,Escherichiacoli、Bacillusamyloliquefaciens、Shigella、Bacillus和Proteusmirabilis5个菌种分布于成年猪肠道的几乎所有节段。在肠道菌群多样性分析中,计算机软件的OTU聚类分析结果可能与16S rDNA全序列测定后的比对结果存在较大的差异。
