气相色谱法测定食用植物油中有机氯类农药残留
2022-10-29任鹏柴艳兵张耀广张洪鑫李兴佳白晓云王鸽董新明
任鹏,柴艳兵,张耀广,张洪鑫,李兴佳,白晓云,王鸽*,董新明
(1.天津食品集团有限公司,天津 300000;2.君乐宝乳业集团农业农村部乳品质量安全控制重点实验室,河北 石家庄 050021;3.天津康复疗养中心,天津 300191)
有机氯农药主要成分为苯和环戊二烯两类[1],用于防治植物疾病与虫害等,其作用明显但具有高毒性、低挥发性、不易降解、高生物积累性等缺点[2],易溶于脂肪和有机溶剂,会通过食物链的放大作用,对人类健康造成危害[3],主要损害中枢神经和肝、肾等。而造成以上损害的主要原因则是中毒,中毒类型分为急性中毒和慢性中毒两种[4-6]。急性中毒症状有头痛、眩晕、恶心、呕吐、流涎、肌无力、肌震颤等,严重者有大汗淋漓、发热、浑身震颤、抽搐、昏迷等症状[7-8]。慢性中毒症状常表现为神经衰弱综合征。慢性中毒的毒理作用主要表现在对神经系统、内分泌系统造成影响并侵害肝脏、肾脏等器官[9-11]。
李曼曼等[12]建立了一种利用QuEChERS(quick、easy、cheap、effective、rugged、safe) 结合气相色谱-三重四极杆串联质谱检测畜禽肉中17种有机氯农药残留的方法。结果显示17种有机氯农药在1 μg/kg~100 μg/kg范围内线性关系良好,检测限为 0.07 μg/kg~0.31μg/kg。王晓春等[13]建立了蔬菜及水果中16种有机氯农药残留的气相色谱检测方法,检出限在0.16 μg/kg~2.90 μg/L范围内,在4种基质中的加标回收率为70.1%~119%,相对标准偏差为0.23%~5.2%。黄小波等[14]建立了QuEChERS方法测定鸡蛋中17种农药的残留,结果表明17种农药在0~0.100 μg/mL范围内,线性关系良好,相关系数R2在0.996 4~0.999 9,加标回收率为76.3%~102%,方法检出限在0.01μg/kg~0.28μg/kg。李妃等[15]建立了海水中16种有机氯农药的气相色谱测定方法,样品经前处理后利用电子俘获检测器(electron capture detector,ECD)进行分析测定,色谱系统采用DB-1701毛细管色谱柱,结果显示在0.75 μg/L~200 μg/L范围内,16种有机氯农药具有良好的线性关系,检出限在0.53 ng/L~2.90 ng/L之间,加标回收率为82.0%~107.9%。李福敏等[16]建立了气相色谱-电子捕获检测器方法检测大米中32种有机氯类农药残留量的分析方法,结果显示32种目标物在优化试验条件下有较好的分离效果,方法检出限为 1.0 μg/kg~20.0 μg/kg,定量限为 2.0 μg/kg~25.0 μg/kg。试验相对标准偏差为 0.19%~8.6%(n=6);张静静[17]通过对吉林省土壤中有机氯类农药的分析,总结出长春市城郊菜地土壤中有机氯农药残留主要为六六六和滴滴涕,两者共占有机氯农药残留总量的88.29%;吉林市城郊菜地土壤中有机氯农药残留主要为六六六和滴滴涕,两者共占有机氯农药残留总量的82.05%;检测城市蔬菜依然有有机氯类农药残留存在。
农药中有机杀虫剂包括有机磷类、有机氯类、氨基甲酸酯类、拟除虫菊酯类、特异性杀虫剂等。我国于1983年已经禁止有机氯类农药的使用与生产[18],但该类农药在土壤与植物中的检出率依然较高。植物是食用植物油的主要原料[19-20],因此食用植物油中存在有机氯农药残留的风险,然而食用植物油和一般食品基质差异较大,主要由脂肪酸甘油酯组成,而大多数农药是脂溶性的,所以检测食用植物油中的农药残留时,必须除去基质中的脂肪、色素及其他相对分子质量较高的干扰物,以免检测仪器受到基质干扰物的污染,导致仪器使用寿命变短。对于食用植物油中残留农药的检测,常用的液-液萃取和固相萃取等前处理步骤繁琐,试剂使用量大,前处理时间长,检测大批量样品时效率低。而QuEChERS法以其快速、简便、价格低廉、有效、可靠和安全的优点在农药残留分析中得到了广泛应用,并已用于蔬菜、大米、水果、茶叶等多种食品基质中。
目前,任雅君等[21]整体介绍了现阶段国内主要的植物油中农药残留的前处理方法以及常用的检测方法(气质联用和液质联用);GB/T 5009.19—2008《食品中有机氯农药多组分残留量的测定》和GB/T 5009.162—2008《动物性食品中有机氯农药和拟除虫菊酯农药多组分残留量的测定》中提出检测食用油采用气相色谱法[22-23],但以上方法的前处理操作均用到大量的石油醚、乙酸乙酯等毒性较大的有机试剂,因此,本研究综合借鉴QuEChERS快速前处理方法,以大豆油为代表性基质,采用N-丙基乙二胺(primary secondary amine,PSA)和C18粉末作为QuEChERS法的吸附剂去除食用植物油基质中的干扰物,结合气相色谱建立快速、高效的食用植物油中18种有机氯农药的检测方法,为今后食用植物油中检测有机氯农药,提供理论依据和技术支撑。
1 材料与方法
1.1 材料与试剂
大豆油(5 L):市售。
18 种农药残留标准品[α-六六六、β-六六六、γ-六六六、δ-六六六、七氯、艾氏剂、氧氯丹、环氧七氯、反-氯丹、α-硫丹、顺-氯丹、狄氏剂、p,p'-DDE,2,2-双(对氯苯基)-1-氯乙烯、β-硫丹、p,p'-DDD、o,p'-DDT、硫丹硫酸盐、p,p'-DDT]:农业部环境保护科研研究所;无水硫酸镁:天津市永大化学试剂有限公司;乙腈(色谱纯):默克化工技术(上海)有限公司;N-丙基乙二胺填料(PSA填料)、C18填料:天津一方科技有限公司;正己烷(色谱纯):北京迪科马科技有限公司。
1.2 仪器与设备
色谱柱(HP-5,柱长 30 m,内径 0.32 mm,膜厚0.25 μm):安捷伦科技(中国)有限公司;气相色谱仪(8890,配电子俘获检测器):岛津仪器苏州有限公司;分析天平(BT25S):德国赛多利斯股份公司;涡旋混合器(IKA-MS3):德国IKA有限公司;旋转蒸发仪(BM510C):日本 Yamato公司;离心机(TGL-20M):上海卢湘仪离心机仪器有限公司。
1.3 试验方法
1.3.1 标准工作液的配制
准确移取18种农药残留标准品(100 μg/mL)1 mL,用正己烷溶解并定容至10 mL,此为标准储备液(10 μg/mL);准确移取标准储备液 1 mL,用正己烷溶解并定容至 10 mL,此为标准中间液(1 μg/mL);准确移取标准中间液1 mL,用正己烷溶解并定容至10 mL,此为标准使用液(100 ng/mL)。
本方法确认所做曲线的工作范围为(5 ng/mL~60 ng/mL),分别吸取标准使用液 50、100、200、300、500、600 μL 用正己烷定容至 1 mL,涡旋混匀,配制成浓度为 5.00、10.00、20.00、30.00、50.00、60.00 ng/mL 的标准工作液。以峰面积为纵坐标,以农药残留各组分含量为横坐标,绘制标准曲线。
1.3.2 仪器条件
检测器类型为ECD,载气:高纯氮气,恒压模式,进样口温度280℃,检测器温度300℃,进样体积1 μL,不分流,电流保持 1.0 nA,程序升温:60 ℃保持1 min,以40℃/min升至170℃,以1.2℃/min升至230℃,再以40℃/min升至280℃,保持5 min。
1.3.3 样品处理
称取2 g样品于50 mL离心管中,加入15 mL乙腈,涡旋混匀,8 000 r/min离心5 min。用吸管将有机相乙腈全部转移到另一离心管中后,重复提取一次,合并提取液。向30 mL提取液中加入10 mL用乙腈饱和的正己烷,除去乙腈中的油脂,涡旋混匀,5 000 r/min离心5 min,取下层乙腈于新的离心管中,加入0.2 g PSA填料,0.2 g C18填料、2 g无水硫酸镁,涡旋混匀,8 000 r/min离心5 min,转移上清液至旋蒸瓶中,旋转蒸干,加入1.00 mL正己烷,在涡旋混合器上充分振荡溶解残留物,经0.22 μm有机系滤膜过滤后,采用气相色谱检测。
1.3.4 色谱条件优化
在保持唯一变量的情况下,固定压力模式、检测器温度、进样体积,电流保持1.0 nA,程序升温:60℃保持1 min,以40℃/min升至170℃,以1.2℃/min升至230℃,再以40℃/min升至280℃,保持5 min,更改分流比、进样口温度进行上机测试。
1.4 数据处理
每组数据均重复3次,采用WPS Office11.1.0.11372-release软件进行数据统计分析,使用Origin2017软件绘制结果图。
2 结果与分析
2.1 色谱条件的优化及标准曲线
进样口温度是样品气化温度,影响化合物气化的关键因素,在保持唯一变量的情况下,不同进样口温度对仪器响应值的影响如图1所示;分流比直接影响进入仪器的气体量及响应值结果,不同分流比对响应值的影响如图2所示。

图1 不同进样口温度对响应值的影响Fig.1 Influence of inlet temperature on response
由图1可知,进样口温度为280℃的18种有机氯农药均有最大响应值,由图2可知,除p,p'-DDT外其余17种有机氯农药在不分流情况下响应值最大。最终确定仪器条件:载气为高纯氮气、恒压模式、进样口温度280℃、检测器温度ECD 300℃、进样体积1 μL、不分流进样、电流1.0 nA。程序升温至60℃保持1 min,以40℃/min升至170℃,以1.2℃/min升至230℃,再以40℃/min升至280℃,保持5 min。
18种有机氯农药分离色谱图如图3所示。
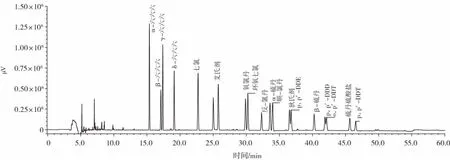
图3 18种有机氯农药色谱图Fig.3 Chromatography of 18 organochlorine pesticides
由图3可知,18种有机氯农药出峰峰形对称,无杂质峰干扰,分离效果较好。所选取浓度为5.00、10.00、20.00、30.00、50.00、60.00 ng/mL,以峰面积为纵坐标,以农药残留各组分含量为横坐标,绘制标准曲线,试验表明,农药残留各组分的含量在0~60 ng/mL内呈线性,具体数值见表1。

表1 农药残留各组分线性分析Table 1 Linearity of each pesticide residue

续表1 农药残留各组分线性情况Continue table 1 Linearity of each pesticide residue
从表1可以看出,18种农药残留混标的各组分线性均大于0.990,符合GB/T27404—2008《实验室质量控制规范食品理化检测》中对校准曲线相关系数的要求。
2.2 前处理方法优化
2.2.1 提取溶剂的确定
分别采用甲醇、乙腈、50%的甲醇乙腈溶液做提取试剂,保持相同提取溶剂体积,用甲醇和甲醇乙腈混合溶液提取时,回收率在45%以下,不满足标准要求,使用乙腈提取样品时回收率达到90%,所以试验选取乙腈作为提取试剂。
2.2.2 除脂试剂的确定
不同除脂试剂的加标回收率见表2。

表2 不同除脂试剂的加标回收率Table 2 Spiked recoveries of different degreasing reagents
乙腈提取大豆油后,部分脂类物质溶于乙腈,造成提取液不能完全蒸发至干,影响定容体积。因正己烷具有除脂效果,故在提取液中加入10 mL正己烷,结果显示使用纯正己烷,除脂效果明显,但试验回收率低,分析原因是正己烷能溶解部分乙腈,尝试加入用乙腈饱和的正己烷,回收率达到标准要求,所以试验选用乙腈饱和的正己烷作为除脂试剂。
2.2.3 正确度与精密度
选择3个不同添加水平进行加标回收率试验,3 个水平的加标量分别为 0.005、0.010、0.020 mg/kg,分别做6个平行,结果见表3。
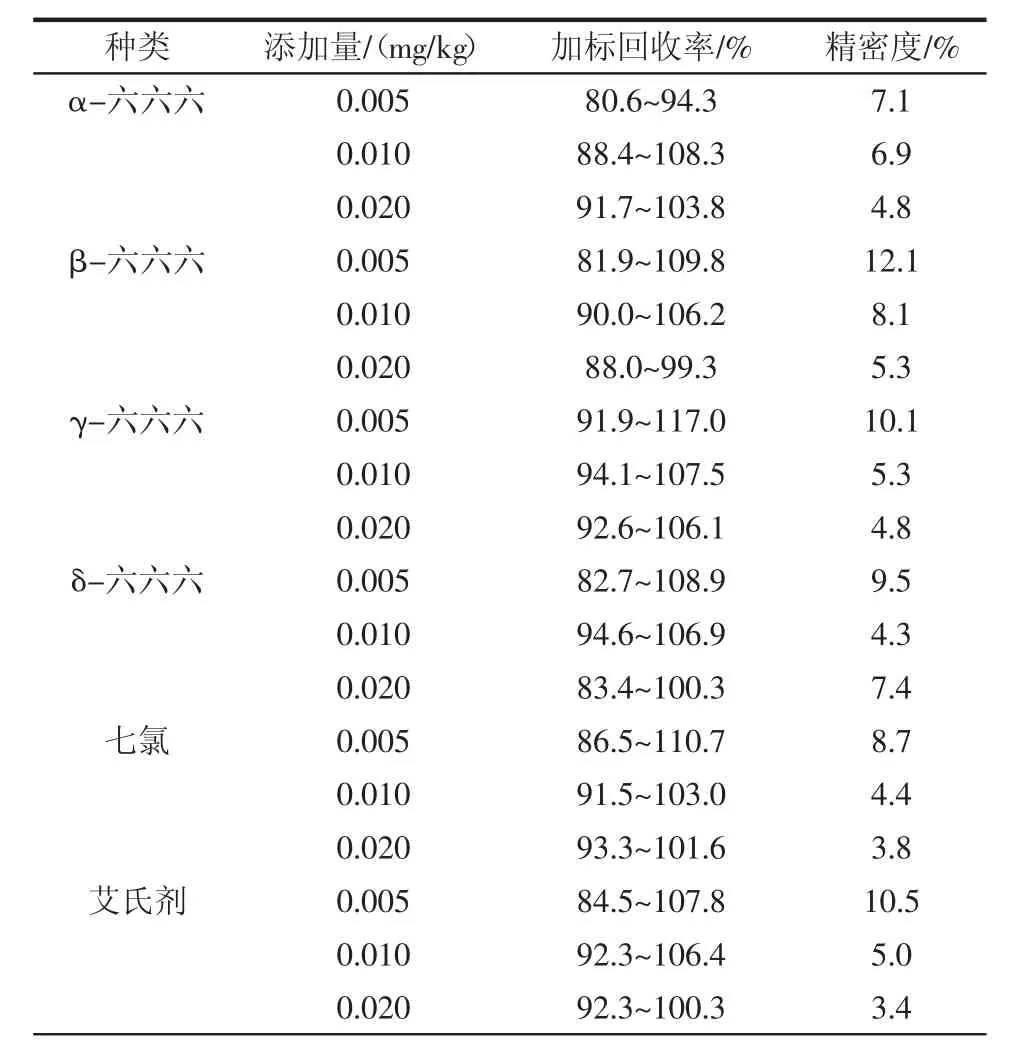
表3 加标回收率Table 3 Spiked recovery
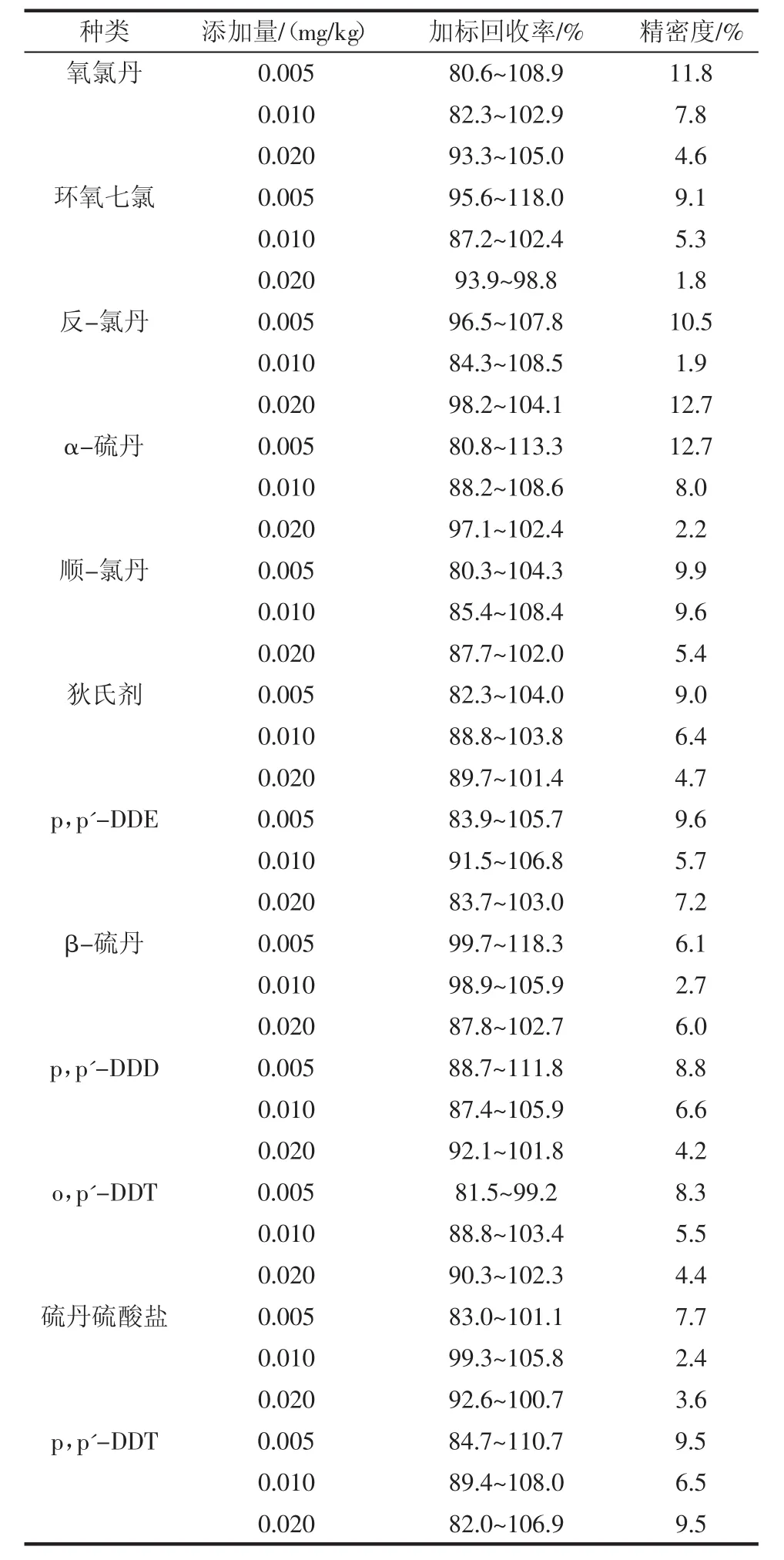
续表3 加标回收率Continue table 3 Spiked recovery
通过表3可以看出,在大豆油基质中,18种有机氯农药加标回收率为80.3%~118.3%。各组分加标回收率可以满足实验室对于大豆油中有机氯农药残留筛查的要求,所得结果的变异系数为1.8%~12.7%,满足GB/T 27404—2008《实验室质量控制规范食品理化检测》中变异系数要求。结果显示该方法有较好的准确度和精密度,可以作为检测大豆油中有机氯农药残留的方法。
3 结论
本文通过优化进样口温度、分流比,选取乙腈作为提取试剂、乙腈饱和的正己烷作为除脂试剂建立了一种气相色谱法检测食用植物油中有机氯农药残留的方法,数据表明该方法提取、除脂效果好,分离度高,适用于脂肪含量高的油类物质。该法操作简单,准确度、灵敏度高,可以满足同时检测食用植物油中18种有机氯农药的要求,为检测食用植物油中其他种类农药残留提供理论依据。
