微波消解—常压消解—电感耦合等离子体质谱法测定植物样中的锗
2022-10-20方晓青胡伟康张飞鸽左天乐
方晓青, 胡伟康, 张飞鸽, 吴 晶, 左天乐
(1.湖北省地质实验测试中心,湖北 武汉 430034; 2.湖北省华祥计量技术有限公司,湖北 武汉 430022)
锗是一种重要的稀有元素,属于人体必需的微量元素之一,具有抗肿瘤、抗衰老、提高免疫力、促进新陈代谢等作用。植物、食品中锗含量的检测早已引起国内外学者的关注,由于其在植物体内含量极低,对检测手段提出了更高的要求。植物样中有机质含量较高,要准确测定锗,消除有机质是首要目标。目前样品前处理技术大致可分为敞开消解和密闭消解两大类,敞开消解包括干灰化消解[1-3]和湿法消解[2-4],密闭消解包括高压密闭罐消解[2]和微波消解[3-4]。
植物中锗的检测方法主要有:原子吸收光谱法[4-5]、分光光度法[6-12]、原子荧光光谱法[1,11-15]和电感耦合等离子体质谱法(ICP-MS)[3,16-17]等。其中,分光光度法使用荧光酮类等显色试剂,所用试剂多有毒有害,且前处理过程繁琐,不适用于批量检测;原子荧光光谱法和原子吸收光谱法线性范围较窄;ICP-MS具有灵敏度高、线性范围宽、分析快速等优点,在日常样品痕量分析中已显示出其独特的优势。马海萍等[16]采用ICP-MS对农产品标准物质进行测定,采用微波消解进行预处理,操作简便,但检出限偏高(0.025 mg/kg),而一般植物样中锗的含量均<0.025 mg/kg,因此该方法不适用于大部分植物样中锗的测定。
本文采用微波消解—常压消解的方式,建立了ICP-MS测定植物样中锗的方法。采用微波消解—常压消解方式提高锗的溶出率;对硫酸、氢氟酸用量及常压消解时间进行优化,进一步改善方法的分析性能;通过数学校正模型扣除仪器测定过程中存在的干扰,保证测试结果准确可靠。该项工作可为相关研究提供数据支持和技术参考。
1 实验部分
1.1 仪器
微波消解仪(MARS6型,美国CEM公司);恒温消解仪(BHW-09C20型,上海博通化学科技有限公司);电感耦合等离子体质谱仪(X2型,美国Thermo Fisher公司)。
1.2 试剂
锗标准储备溶液(锗浓度1 000 mg/L,国家有色金属及电子材料分析测试中心);锗标准溶液(采用锗标准储备液逐级稀释成100 ng/mL);硝酸(上海傲班科技有限公司,优级纯);氢氟酸(北京化工厂,优级纯);硫酸(国药集团,优级纯);过氧化氢(国药集团,CMOS级)。
1.3 仪器工作条件
仪器工作条件见表1和表2。

表1 微波消解实验条件
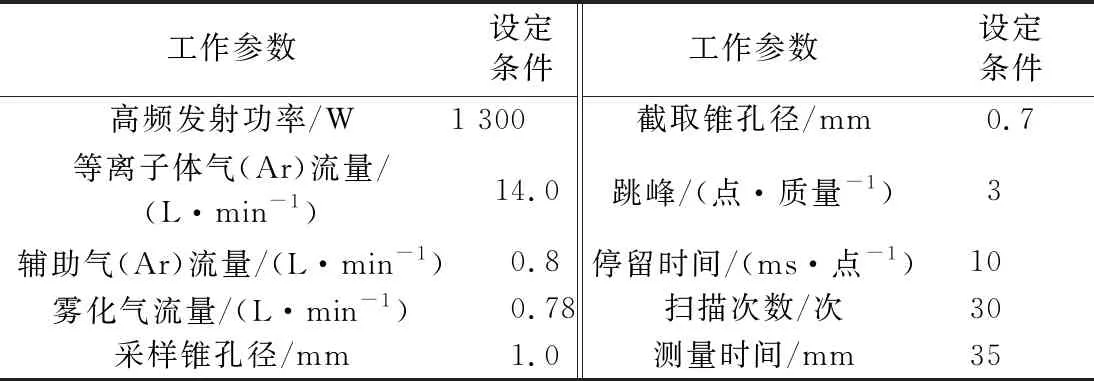
表2 ICP-MS工作条件
1.4 标准曲线绘制
锗工作曲线溶液的配制:吸取适量的锗标准溶液,以2%硝酸配制成标准中间液1 mg/L,用2%硝酸溶液定容至刻度,摇匀。使用时分别移取适量的标液并用2%硝酸配制成锗浓度分别为0.1、0.5、1.0、2.0、5.0 μg/L的标准系列工作溶液。将电感耦合等离子体质谱仪开机调试好,稳定30 min后,由低到高依次测定标准系列工作溶液,绘制标准曲线。
1.5 样品处理
准确称取0.500 0 g(±0.000 5 g,含水分较多的样品可适当增加取样量至1 g)样品于微波消解聚四氟乙烯内罐中(糖分或淀粉含量高的样品先在电热板上低温加热去除有机质),加入6 mL硝酸和1 mL过氧化氢溶液,盖上内盖,旋紧罐盖,按照微波消解仪标准操作步骤进行消解。冷却后取出,缓慢打开罐盖排气,用少量水冲洗内盖,将消解罐中溶液转移至聚四氟乙烯坩埚中,加1 mL硫酸和3 mL氢氟酸,盖上坩埚盖,置于电热板上,升温至100℃。消解3 h后揭盖,继续升温至260℃赶酸,待浓烟冒尽且电热板降温至160℃,加2 mL硝酸提取,赶酸至<0.5 mL,用蒸馏水定容至10 mL,混匀待测,同时做空白试验。
2 结果与讨论
2.1 植物样前处理研究与优化
2.1.1微波消解体系
常用的消解试剂有硝酸、硫酸、盐酸、过氧化氢、高氯酸、氢氟酸等,在温度高于86℃和盐酸存在条件下锗(Ⅳ)与盐酸形成易挥发的四氯化锗而损失[18],因此对应使用的酸体系有硝酸—硫酸、硝酸—过氧化氢等。考虑到消解体系是密闭状态,所以尽量不选择高氯酸,最终选择硝酸作为主要消解试剂,加入过氧化氢加快消解时间和提高消解效果。通过多次试验确定,在加入6 mL硝酸和1 mL过氧化氢的试剂后,样品消解的效果最佳。
选取GBW10023(紫菜)、GBW10052(绿茶)、GBW10045(湖南大米)和GBW10048(芹菜)等4种涵盖水生植物、茶叶、粮食、蔬菜的生物国家一级标准物质进行条件实验。称取0.500 0 g标准物质,加入6 mL硝酸和1 mL过氧化氢,按照表1的微波消解实验条件消解后,置于电热板上赶酸至小体积后定容至10 mL,进行ICP-MS上机测量,结果如表3所示。
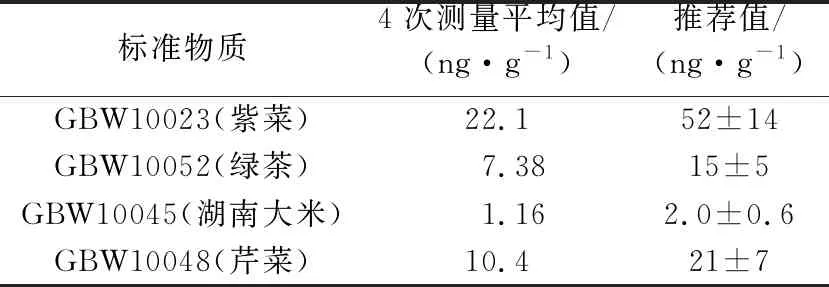
表3 微波消解后标准物质测定值
结果表明,微波消解后,标准物质测定值仅为推荐值的1/2左右,且GBW10023(紫菜)和GBW10052(绿茶)样品消解后有不溶沉淀物,可能是有机质未消解完全或者样品中含有二氧化硅,导致锗的溶出率偏低,还需用强氧化性的硫酸和氢氟酸进行进一步消解。
此时Label[j]数组记录了所有共同连通域的标号,但共同连通域的标号是断序的,不利于连通域的个数统计,因此还需要按照其出现的次序对其进行排序并且输出连通域的个数。
2.1.2微波消解—常压消解体系
为了验证微波消解—常压消解加酸种类对锗溶出率的影响,同样选取GBW10023(紫菜)、GBW10052(绿茶)、GBW10045(湖南大米)和GBW10048(芹菜)等4种不同类型的生物国家一级标准物质进行条件实验。
(1) 硫酸加入量的优化。称取样品0.500 0 g,加入6 mL硝酸和1 mL过氧化氢,按照表1的微波消解实验条件消解后,将溶液转移至聚四氟乙烯坩埚中,分别加入不同体积的硫酸(0、1、2和3 mL),盖上坩埚盖,100℃消解3 h后升温至260℃赶酸,待白烟冒尽且电热板温度降至160℃,加2 mL硝酸,赶酸至硝酸体积<0.5 mL后定容至10 mL,进行ICP-MS上机测量。比较加入不同体积硫酸消解后的标准物质的测量值,结果如图1所示。

图1 不同体积硫酸加入的条件实验结果
结果表明,加入硫酸对测量结果的影响非常明显,4种不同类型标准物质的测量值均有明显提高,但硫酸量加到>1 mL之后,锗含量的变化并不明显,因此采用硫酸的加入量为1 mL。标准物质测定值与其推荐值相比,还是有明显差别,但硫酸的加入提高了锗的溶出率。
(2)氢氟酸加入量的优化。称取样品0.500 0 g,加入6 mL硝酸和1 mL过氧化氢,按照表1的微波消解实验条件消解后,将溶液转移至聚四氟乙烯坩埚中,均加入1 mL硫酸,并分别加入不同体积的氢氟酸(0、1、3和5 mL),盖上坩埚盖,100℃消解3 h后升温至260℃赶酸,待白烟冒尽且电热板温度降至160℃,加2 mL硝酸,赶酸至硝酸体积<0.5 mL后定容至10 mL,进行ICP-MS上机测量。比较加入不同体积氢氟酸消解后的标准物质的测量值,结果如图2所示。
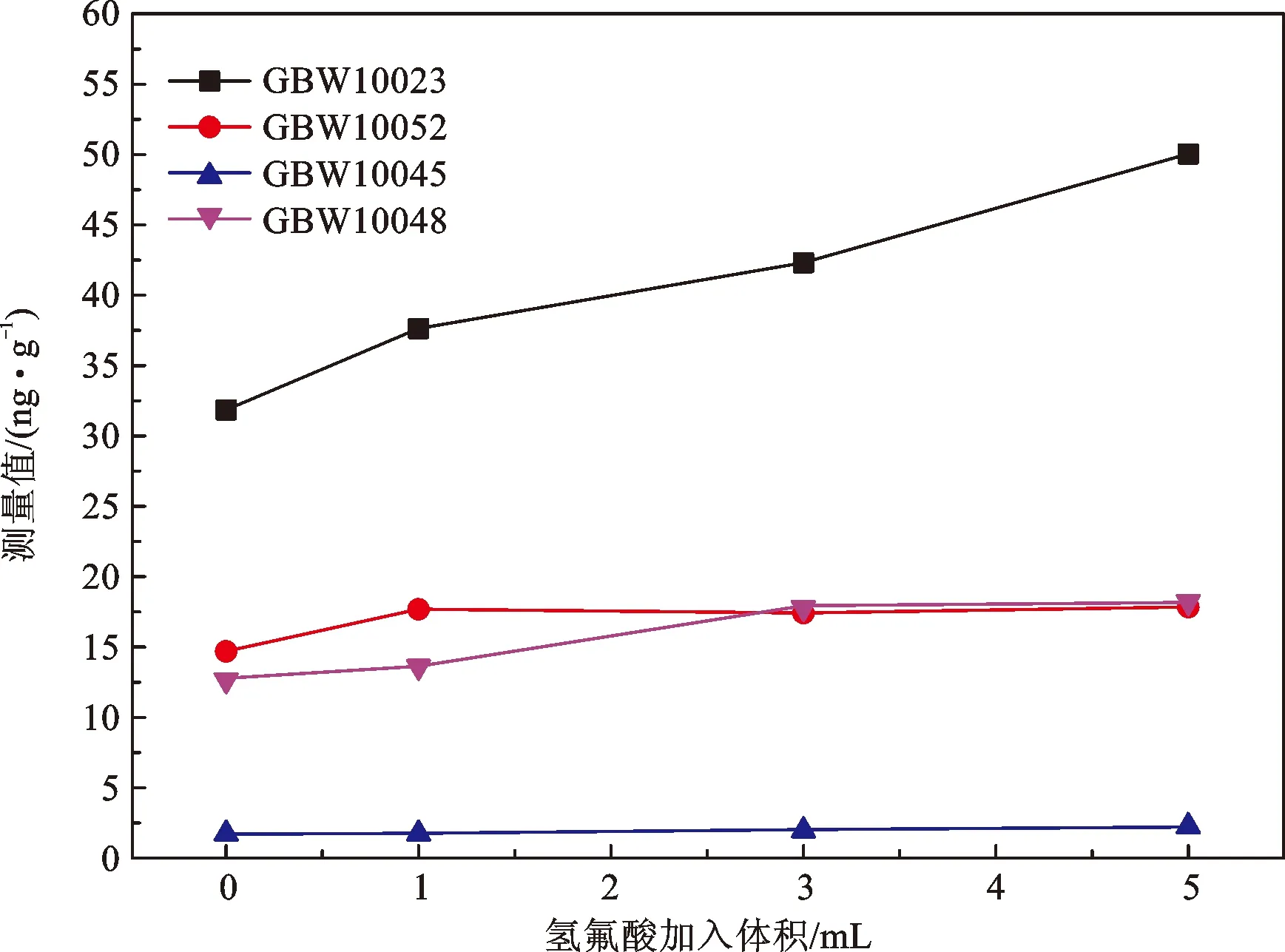
图2 不同体积氢氟酸加入的条件实验结果
结果表明,随着氢氟酸的加入,标准物质的测量值均有明显提高。加入1 mL氢氟酸后,随着氢氟酸体积的增加,GBW10052(绿茶)、GBW10045(湖南大米)和GBW10048(芹菜)的测量值没有明显提高,而GBW10023(紫菜)的测量值增加明显,氢氟酸越多,对紫菜的消解效率越高,可能是因为紫菜中硅的含量较高(推荐值为(0.83±0.16)%)。对于硅含量较高的植物样,可适当增加氢氟酸体积至5 mL,其他样品仅需加3 mL。考虑到加入氢氟酸的体积越大,空白越高,从而影响样品的检出限,当氢氟酸的加入体积为3 mL时,紫菜的锗的测量值也在推荐值的不确定度范围内,因此采用氢氟酸加入量为3 mL。
(3) 常压消解时间的优化。常压消解时间的长短也会影响样品中锗的消解效果。称取样品0.500 0 g,加入6 mL硝酸和1 mL过氧化氢,按照表1的微波消解实验条件消解后,将溶液转移至聚四氟乙烯坩埚中,均加入1 mL硫酸和3 mL氢氟酸后盖上坩埚盖,在100℃分别消解1、2、3和4 h后升温至260℃赶酸,待白烟冒尽且电热板温度降至160℃,加2 mL硝酸,赶酸至硝酸体积<0.5 mL后定容至10 mL,进行ICP-MS上机测量。比较不同常压消解时间消解后的标准物质的测量值,结果如图3所示。
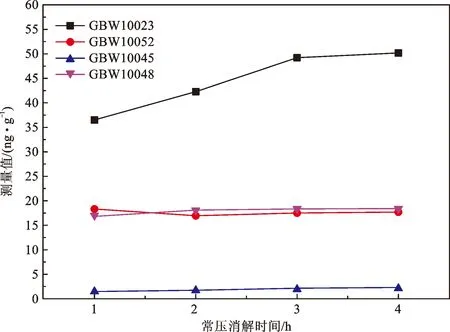
图3 常压消解时间的条件实验结果
结果表明,常压消解时间对GBW10052(绿茶)、GBW10045(湖南大米)和GBW10048(芹菜)的测量值没有明显影响,而GBW10023(紫菜)的测量值提高明显,消解时间越长,紫菜的消解效率越高。为了保证紫菜类植物样的锗的消解效率,选取常压消解时间为3 h进行前处理。
2.2 干扰和消除
2.2.1质谱型干扰
在ICP-MS测量中,质谱干扰主要有同量异位素叠加干扰、难熔氧化物离子干扰、多原子复合离子干扰及双电荷离子干扰等,其中质谱峰叠加、多原子复合离子所形成的干扰较为严重[19]。选择的最佳ICP-MS工作条件见表2,在此工作条件下,铈氧化物的产率<3%,138Ba的双电荷产率<3%。针对质谱型干扰,可以通过两种方法来克服:①选择测量元素的同位素;②通过校正方程进行校正。
(1) 锗同位素的选择。尽量避免选择有大量干扰元素存在及干扰机理复杂的测量同位素,这样可以有效降低部分质谱干扰。锗主要有70Ge、72Ge、74Ge等3个同位素,在测定锗时,主要受到70Zn和140Ce2+对70Ge、144Sm2+和144Nd2+对72Ge、148Sm2+和148Nd2+对74Ge的干扰。植物样中钐和钕的含量很低,可选择74Ge作为测量同位素,干扰相对较小且74Ge的灵敏度也较高。
(2) 干扰校正方程。对选择同位素仍然不能有效避免的质谱干扰,可以通过校正方程进行数学模式干扰校正。校正方程中的干扰系数用钕纯标准溶液测定和计算得出。配制钕浓度分别为100、500、1 000 μg/L的单标溶液,按照表2的ICP-MS工作条件进行检测,得到不同浓度下74Ge、146Nd和148Nd的强度响应值,计算出钕对锗的干扰系数,如表4所示。
由表4计算出146Nd对74Ge的干扰系数为0.001 4,

表4 钕对74Ge的干扰情况
则测定锗时,选定的测定同位素的校正方程为:
C74Ge,校=C74Ge,测-0.001 4C146Nd
式中:C74Ge,校为锗的校正结果;C74Ge,测为锗的测定结果;C146Nd为钕的测定结果。
2.2.2非质谱型干扰
非质谱型干扰主要包括基体抑制干扰、空间电荷效应干扰、物理效应干扰等[20]。非质谱型干扰程度与样品基体性质有关,通过内标法、仪器条件最佳化等措施可以消除。内标元素的选择原则:被测溶液中不含所选内标元素且该元素的测定干扰较少,质量数与被测元素比较接近。基于此,本方法选用103Rh作为内标元素,其浓度为2.0 μg/L。
2.3 检出限
按照本方法进行空白样品制备,测试12份空白样品,以3倍的标准偏差计算检出限,用4倍检出限来估算测定下限,得到锗的检出限为0.59 ng/g,测定下限为2.36 ng/g。
2.4 精密度与准确度
按照本方法选取了6个不同类型的生物国家一级标准物质进行8次测定,并计算精密度和准确度,结果如表5所示。
由表5可看出,测量平均值均在推荐值的不确定范围内,标准物质的精密度均<15%。
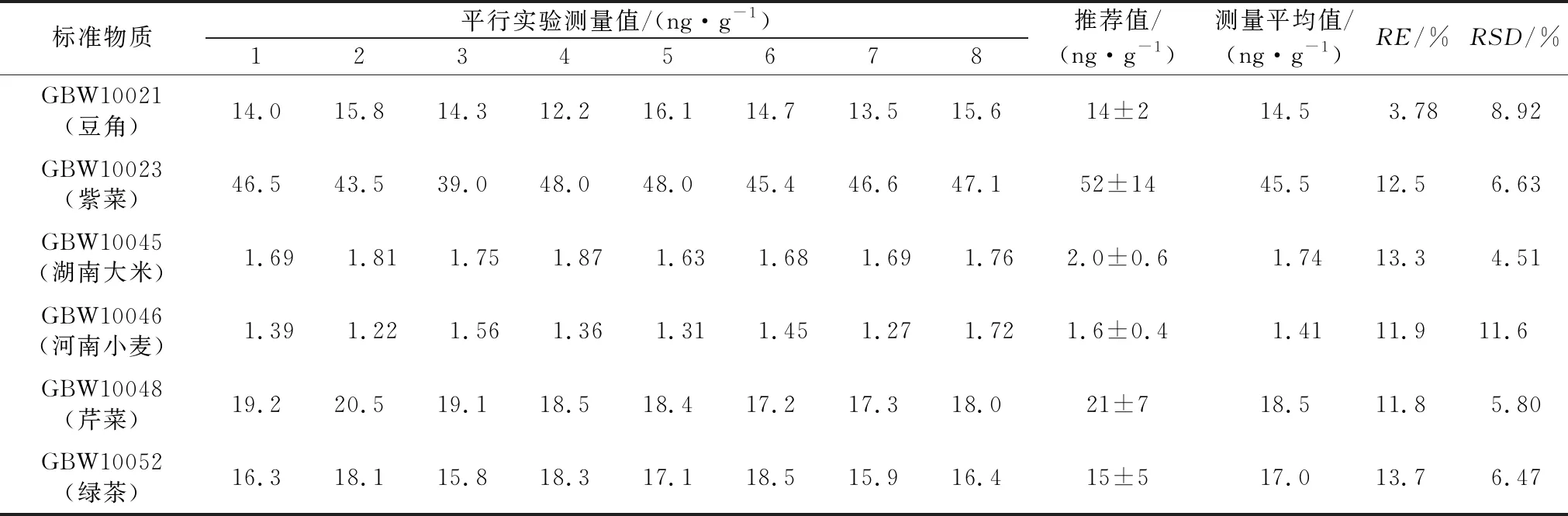
表5 方法准确度和精密度实验结果
3 结论
本文采用微波消解—常压消解前处理方式和ICP-MS测定了生物国家一级标准物质中锗的含量,对比研究了不同的硫酸、氢氟酸用量及常压消解时间对标准物质的消解效果,探究了最优的样品处理方法。结果表明,锗的检出限为0.59 ng/g,测定下限为2.36 ng/g,相对误差(RE)为3.78%~13.7%,相对标准偏差(RSD,n=8)为4.51%~11.6%。该方法检出限低,精密度、准确度良好,都能满足分析要求,适用于植物样中痕量锗的测定。
